Aromaticidade
Aromaticidade é uma propriedade química na qual certos sistemas são mais estáveis do que deveriam ser, são mais simétricos do que se presumia e apresentam característica magnética peculiar. Na verdade os especialistas sabem caracterizar e reconhecer sistemas aromáticos, mas estão longe de um consenso sobre qual a sua origem. Usualmente um anel conjugado de ligações insaturadas, pares de elétron isolados, ou orbitais vazios exibem uma estabilização mais forte do que a esperada devido apenas à conjugação. Também pode ser considerada como uma manifestação de deslocalização cíclica e de ressonância.[1][2][3]
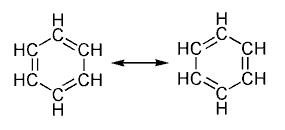
Geralmente considera-se que ela ocorre devido aos elétrons estarem livres para circular por arranjos circulares de átomos, os quais se ligam um ao outro por uma ligação simples e uma ligação dupla alternadamente. Essas ligações são comumente vistas de acordo com o modelo de anéis aromáticos desenvolvido por Kekulé. O modelo de Kekulé considera que as ligações consistem de um híbrido de ligações simples e dupla e que todas as ligações no anel são idênticas. Para o benzeno, o modelo consiste em duas formas de ressonância, que correspondem a ligações simples e duplas trocando de lugar entre si. O benzeno é uma molécula mais estável do que seria esperado, sem contar-se com deslocalização da carga.
Índice
1 Teoria
2 Características dos Compostos aromáticos
3 Importância dos compostos aromáticos
3.1 Critérios de Aromaticidade
3.1.1 Critérios Geométricos
3.1.2 Critérios Energéticos
3.1.3 Critérios Magnéticos
3.1.3.1 Susceptibilidade Magnética (χ) e Exaltação da Susceptibilidade (Λ)
3.1.3.2 Índice de aromaticidade NICS (Nucleus Independent Chemical Shift)
4 Tipos de compostos aromáticos
4.1 Heterocíclicos
4.2 Policíclicos
4.3 Substituídos Aromáticos
4.4 Compostos aromáticos atípicos
4.5 Anulenos
5 Efeito Mills-Nixon
6 Aromaticidade de Superátomos
7 Aromaticidade por Orbitais Moleculares
7.1 Orbital Molecular da Estrutura do Benzeno
7.2 Espécies Aromáticas Carregadas
8 Aromaticidade pela teoria de Ligação de Valência
9 Aromaticidade pela teoria de Átomos em Moléculas
10 Antiaromaticidade
10.1 Cátion ciclopentadienil
10.2 Cátions indenil e fluorenil
10.3 Dicátions Bisfluorenila
11 Novas Teorias
11.1 DI (delocalization index)
11.2 D3BIA (density, degeneracy, delocalization-based Index of Aromaticity)
11.3 Teoria isoeletrônica complexa (Cplex-isoelectronic theory):[71]
11.3.1 Teoria do funcional da densidade
12 Ver também
13 Referências
Teoria |

Representação alternativa
Como é padrão para diagramas de ressonância, uma seta de duplo sentido é usada para indicar que as duas estruturas não são entidades distintas, mas meramente possibilidades hipotéticas. Nem é uma representação precisa de compostos reais, o que é melhor representada por um híbrido (média) dessas estruturas, que pode ser visto à direita. A ligação C=C é menor do que uma ligação C-C, mas o benzeno é perfeitamente hexagonal - todas as seis ligações carbono-carbono tem a mesma distância, intermediária entre aquela de uma ligação simples e uma dupla.
A melhor representação é aquela da ligação π circular (círculo interno de Armstrong), em que a densidade de elétrons é distribuída através de uma ligação π acima e abaixo do anel. Este modelo representa mais corretamente a localização da densidade de elétrons no anel aromático.
As ligações simples são formadas de acordo com os elétrons entre os núcleos de carbono - estas são chamadas ligações σ. Ligações duplas consistem de uma ligação σ e uma ligação π. As ligações π são formadas de sobreposição de orbitais atômicos p acima e abaixo do plano do anel. O diagrama a seguir mostra as posições destes orbitais p:

Por estarem fora do plano dos átomos, estes orbitais podem interagir uns com os outros livremente, e tornam-se deslocalizados. Isto significa que ao invés de estarem amarrados a um átomo de carbono, cada elétron é compartilhado por todos os seis no anel. Assim, não há elétrons o suficiente para formar ligações duplas em todos os átomos de carbono, mas os elétrons "extra" reforçam todas as ligações no anel igualmente. O orbital molecular resultante tem simetria π.
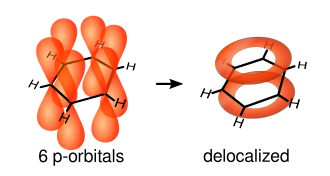
Características dos Compostos aromáticos |
Um composto aromático (ou composto de arilo ou arila, ou ainda, "arilcomposto") contém um conjunto de átomos ligados covalentemente com características específicas:
- Um sistema conjugados de π deslocalizados, mais comumente um arranjo de alternância de ligações simples e duplas.
- Estrutura coplanar, com todos os átomos contribuintes no mesmo plano.
- Átomos contribuintes dispostos em uma ou mais anéis.
- Um certo número de elétrons π deslocalizados que é um número par, mas não um múltiplo de 4. Isto é, um número de 4n + 2 = n° elétrons π e/ou pares livres, onde n = 0, 1, 2, 3, e assim por diante. Isso é conhecido como regra de Hückel.
Considerando que o benzeno é aromático (6 elétrons, a partir de 3 duplas ligações), o ciclobutadieno não é, pois o número de elétrons π deslocalizados é 4, o que evidentemente é um múltiplo de 4. O íon ciclobutadienido (um íon de carga 2 -), no entanto, é aromático (6 elétrons). Um átomo de um sistema aromático pode ter outros elétrons que não fazem parte do sistema e, portanto, ignorado pela regra 4n + 2. No furano, o átomo de oxigênio é hibridizado sp². Um par de elétrons π não está no sistema e os outros estão no plano do anel (análoga à ligação C-H em outras posições). Há 6 elétrons π, então o furano é um composto aromático.
Moléculas aromáticas normalmente exibem uma estabilidade química ampliada em comparação às moléculas semelhantes não aromáticas. Uma molécula que pode ser aromática tenderá a alterar a sua estrutura eletrônica e conformacional para estar nesta situação. Estas estabilidade extra muda a química da molécula . Compostos aromáticos sofrem reações de substituição eletrofílica aromática e substituição nucleofílica aromática, mas não reações de adição eletrofílica como acontece com ligações carbono-carbono duplas.
Muitos dos primeiros exemplos conhecidos de compostos aromáticos, como benzeno e tolueno, tem cheiro agradável distintivo. Esta propriedade levou ao termo aromático para esta classe de compostos, e daí o termo "aromaticidade" para a propriedade eletrônica posteriormente descoberta.
Os elétrons π que circulam em uma molécula aromática produzem correntes de anel que se opõem ao campo magnético aplicado em RMN. O sinal de RMN de prótons não planos de um anel aromático é mantido substancialmente mais "campo abaixo" (sentido do campo, relacionado à corrente de anel) do que aqueles de carbonos sp² não aromáticos. Esta é uma importante forma de detecção de aromaticidade. Pelo mesmo mecanismo, os sinais de prótons localizados próximos do eixo do anel é mantido "campo acima".
Moléculas aromáticas são capazes de interagir umas com as outras no assim chamado empilhamento π-π: os sistemas π formam dois anéis paralelos com sobreposição em orientação "face-a-face". Moléculas aromáticas também são capazes de interagir umas com as outras em uma orientação "ponta-a-face": uma pequena carga positiva dos substituintes no anel de átomos de uma molécula são atraídos para uma carga ligeiramente negativa do sistema aromático em uma outra molécula.
Moléculas monocíclicas contendo 4n elétrons π planares são chamadas antiaromáticas e são em geral, desestabilizadas. Moléculas antiaromáticas também tenderão a alterar a sua estrutura eletrônica e conformacional para evitar esta situação, tornando-se não aromáticas. Por exemplo, ciclo-octatetraeno (COT) distorce-se fora da planaridade, quebrando uma sobreposição entre as ligações π duplas adjacentes. Relativamente recentemente, descobriu-se que o ciclobutadieno adota uma configuração assimétrica retangular na qual ligações simples e duplas alternam-se de fato, não há ressonância e as ligações simples são nitidamente mais longas do que as ligações duplas, reduzindo-se uma desfavorável sobreposição de orbitais p. Consequentemente, ciclobutadieno é não-aromático, com uma tensão da configuração assimétrica que supera uma desestabilização antiaromática que perturba uma configuração quadrada e simétrica.
Importância dos compostos aromáticos |
Os compostos aromáticos são importantes na indústria. Hidrocarbonetos aromáticos chaves de interesse comercial são benzeno, tolueno, orto-xileno e para-xileno. Cerca de 35 milhões de toneladas são produzidas em todo o mundo a cada ano. Eles são extraídos de misturas complexas obtidas pelo refino de petróleo ou pela destilação do alcatrão de carvão, e são utilizados para produzir uma gama de produtos químicos e polímero s importantes, incluindo estireno, fenol, anilina, poliéster e nylon.
Outros compostos aromáticos desempenham papéis-chave na bioquímica de todos os seres vivos. Três aminoácidos aromáticos, fenilalanina, triptofano e tirosina, cada um servindo como um dos 20 blocos básicos de construção das proteínas. Além disso, todos os 5 nucleotídeos (adenina, timina, citosina, guanina e uracila) que compõem a sequência do código genético (DNA e RNA) são purinas ou pirimidinas aromáticas. Bem como a molécula hemo que contém um sistema aromático com 22 elétrons π. A clorofila tem também um sistema aromático semelhante.
Critérios de Aromaticidade |
O conceito de aromaticidade é historicamente citado e utilizado na literatura[4] , sendo este caracterizado por critérios teóricos que são divididos em: geométricos, energéticos e magnéticos. Os critérios geométricos se baseiam nas informações estruturais relacionadas ao comprimento das ligações que indicam estruturas deslocalizadas. Os critérios energéticos se baseiam na estabilidade termodinâmica. E os critérios magnéticos se baseiam nos níveis de energia, na distribuição eletrônica e na polarizabilidade.[5]
O estudo entre a relação de cada critério teórico apresentado pode apresentar variações de acordo com a classe de compostos a serem analisados. Desta forma, para um melhor entendimento do conceito de aromaticidade deve-se utilizar o maior número de diferentes critérios considerando as limitações de cada um.[6]
Critérios Geométricos |
O conceito de aromaticidade leva em consideração os comprimentos das ligações em função da deslocalização de elétrons em estruturas cíclicas. Com isso, a determinação do comprimento de ligação serve como ferramenta de estudo para interpretação e quantificação de tal deslocalização.
O benzeno é um exemplo de uma molécula hexagonal com todos os lados de comprimentos idênticos, o que é contrário ao esperado, uma vez que esta deveria possuir dois tipos de comprimentos de ligações diferentes. Entretanto, o comprimento de todas as ligações C-C observadas para essa molécula é de 1,39 Å, que é inferior ao comprimento de uma ligação simples (1,54 Å) e superior ao de ligações duplas (1,34 Å). A explicação para esse fenômeno se encontra na deslocalização dos elétrons das ligações duplas do benzeno por toda sua estrutura, gerando um comprimento intermediário para as ligações C-C, o que está associado à aromaticidade da molécula. Entretanto, o mesmo comportamento não é observado na análise dos comprimentos de ligação do ciclobutadieno, pois esta molécula possui formato retangular e não quadricular. Ou seja, compostos antiaromáticos possuem elétrons localizados e geralmente possuem ligações simples e duplas alternadas com comprimentos diferentes em no mínimo 0,2 Å.[7]
Como foram observados comportamentos diferentes entre estruturas cíclicas com ligações duplas conjugadas, modelos matemáticos foram sugeridos para caracterização da aromaticidade em função da variação do comprimento da ligação. Krygowski e colaboradores [8] desenvolveram um modelo chamado HOMA (Oscilador Harmônico para a aromaticidade), que considera tanto a alternância de comprimentos de ligações (GEO) como o elongamento das ligações (EN).
O índice HOMA descreve o decréscimo da aromaticidade do sistema. Dessa forma, quanto menor a aromaticidade do composto, menor é o índice HOMA, cujo valor máximo ideal é 1.
HOMA = 1 — [α (Ropt — Rav)² + (α/n) Σ(Rav — Ri)²] = 1 — EN — GEO (1)
Para Rav = Média dos comprimentos de ligação. Obtido do composto cuja aromaticidade será avaliada.
Ropt = Raio ótimo. Obtido da média de comprimentos de ligação de um hidrocarboneto alifático como o butadieno.
Ri = Comprimentos de ligação individuais.
n = Número de ligações.
α = Constante empírica que fornece HOMA = 0 para estruturas de Kekulé hipotéticas, e HOMA = 1 para estruturas com todos os comprimentos de ligação iguais a Ropt.
O primeiro termo da equação 1 (EN), que corresponde ao desvio do comprimento ótimo de ligação, contribui para a energia associada com efeitos de alongamento do comprimento de ligação, e o segundo termo (GEO) reflete o desvio do comprimento médio de ligação, contribuindo para a alternância dos comprimentos de ligação.
Quanto maior o termo de elongação (EN) e de alternância (GEO), menor será o índice HOMA, e, portanto, o sistema será menos aromático.[9]
Dessa forma, o modelo Oscilador Harmônico para a Aromaticidade permite medir a aromaticidade de cada um dos anéis em policiclos, ou seja, a aromaticidade local.
Critérios Energéticos |
Diferentemente dos critérios geométricos, os critérios energéticos consideram a aromaticidade de todo o composto. Para avaliar a aromaticidade de um composto, pode-se determinar a extensão de sua estabilização termodinâmica em termos da energia deslocalizada pelo sistema, que está relacionada às energias de ressonância. Os compostos aromáticos apresentam estabilização energética quando comparados com compostos que não apresentam aromaticidade.
Uma ligação dupla do ciclo-hexano possui entalpia de hidrogenação de 25,6 kcal/mol, e um ciclo-hexa-1,3-dieno possui entalpia de hidrogenação de 54,1 kcal/mol. A entalpia de hidrogenação prevista para um ciclo-hexatrieno hipotético seria de 85,8 kcal/mol, entretanto, o valor observado é menos exotérmico do que o valor previsto, ou seja, de 49,8 kcal/mol. Portanto, o benzeno é mais estável em 36,0 kcal/mol do que um ciclo-hexatrieno hipotético. Essa estabilidade adicional é conhecida como energia de ressonância do benzeno (energia de estabilização aromática).[10] Elétrons compartilhados por somente dois núcleos constituem ligações químicas localizadas, enquanto elétrons compartilhados por mais de dois núcleos constituem ligações deslocalizadas. Para avaliar a estabilidade termodinâmica existente em compostos cujos elétrons se encontram delocalizados, ou seja, aromáticos, pode ser analisada em termos de energia de ressonância (ER). Essa energia pode ser usada para comparar sistemas relacionados, não sendo uma grandeza física mensurável. No entanto, há modelos para se determinar aproximações da extensão da estabilização resultante da deslocalização aromática.
O primeiro critério energético foi determinado por Pauling[11] e Kistiakowsky[12]. Eles propuseram medidas experimentais das energias de ressonância para medir a aromaticidade. É possível calcular a energia de ressonância a partir da diferença entre as energias HOMO e LUMO, ou seja, os orbitais de fronteira.
Fukunaga e Haddon[13] analisaram a relação entre as diferenças de energia entre HOMO e LUMO e as energias de ressonância. Foi observado que quanto maior a estabilização aromática de um composto, maior a diferença entre os orbitais de fronteira. Os autores deduziram a seguinte expressão:
RE = - [(πρrs)²/24] (εLUMO — εHOMO) (2)
A diferença de energia entre os orbitais de fronteira de um composto é expressa por (εLUMO — εHOMO). (πρrs) é a média das ordens de ligação do sistema.
Quanto maior a aromaticidade do sistema, maior o valor da energia de ressonância (RE).
Os valores de RE obtidos a partir da equação 2 são teóricos, devendo ser considerados como índices de aromaticidade, ou seja, não reproduzem necessariamente os valores obtidos experimentalmente.
Uma outra forma de se determinar as energias de ressonância são as reações isodésmicas[14] e homodesmóticas[15]. Reações isodésmicas são reações hipotéticas não necessariamente executáveis em laboratórios. Nesse tipo de reação, pode-se analisar alguns efeitos da estrutura eletrônica, como as tensões anelares e a aromaticidade nos compostos. É feita a otimização dos compostos por cálculos teóricos (com funções de base e métodos que os descrevam), e assim, se faz um cálculo termoquímico da energia da reação.
Nas reações isodésmicas, deve ser mantido o mesmo número de ligações e de átomos nos reagentes e produtos. Enquanto nas reações homodesmóticas, além das condições citadas para as reações isodésmicas, deve também ser mantido o número de átomos com suas hibridizações correspondentes.
A partir deste modelo teórico, é possível prever a variação de entalpia de diversas reações e comparar os valores obtidos com valores experimentais correspondentes.
Critérios Magnéticos |
Compostos aromáticos são conhecidos por apresentarem correntes diamagnéticas quando submetidos a um campo magnético externo. Em 1936, Pauling demonstrou que, quando estão sob ação de um campo magnético, os elétrons pz dos carbonos do composto aromático se movimentam livremente entre os carbonos adjacentes.[16] O movimento eletrônico promove a formação de correntes de anel, que influenciam nas propriedades magnéticas dos compostos.
Susceptibilidade Magnética (χ) e Exaltação da Susceptibilidade (Λ) |
A susceptibilidade magnética descreve o comportamento de um composto ao ser exposto a um campo magnético externo. Pascal[17] verificou que esta era uma propriedade aditiva dos átomos e das ligações pertencentes aos compostos. Compostos aromáticos possuem um aumento desta susceptibilidade se comparar com compostos isentos dos efeitos de aromaticidade. Em função disso, compostos aromáticos são considerados diamagnéticos e possuem uma “exaltação da susceptibilidade magnética”. Compostos aromáticos são diamagnéticos por não apresentarem magnetização espontânea a campo nulo, mas apresentarem magnetização contrária a um campo aplicado, ou seja, susceptibilidade magnética negativa. Enquanto compostos que não possuem magnetização a campo zero, mas que apresentam magnetização no mesmo sentido de um campo aplicado, possuem uma susceptibilidade magnética positiva, e por isso chamados de paramagnéticos.
M = χ Bo (3)
Para M = Magnetização
χ = Susceptibilidade magnética molar
Bo = Campo magnético aplicado
Compostos com valores de χ inferiores a zero são diamagnéticos, enquanto compostos com χ superiores a zero são paramagnéticos.
A susceptibilidade magnética pode ser descrita como uma forma de se estimar em quanto os elétrons de um composto são capazes de induzir um campo magnético no mesmo sentido ou não do campo aplicado. Portanto, compostos aromáticos possuem uma susceptibilidade alta, uma vez que podem induzir campos magnéticos consideráveis de forma a interferir nas freqüências de ressonância de átomos ligados ao anel aromático.[18]
Em função da predominância dos efeitos diamagnéticos em compostos aromáticos, os valores absolutos de susceptibilidade magnética podem ser usados como parâmetro para medição da aromaticidade. No entanto, a exaltação da susceptibilidade é a grandeza mais utilizada para quantificar a aromaticidade de um composto. Esta grandeza pode ser calculada pela diferença entre a susceptibilidade medida experimentalmente (χm) e a susceptibilidade estimada de uma substância referência (χa)[19], isto é, que não seja aromática.
Λ = χm - χa
Compostos com valores de Λ inferiores a zero são aromáticos enquanto compostos com valores de Λ superiores a zero são antiaromáticos.
Índice de aromaticidade NICS (Nucleus Independent Chemical Shift) |
O Critério NICs é um dos métodos mais recentes para se medir a aromaticidade se baseando em critérios magnéticos. Nesse critério é proposto usar blindagens magnéticas absolutas, e por isso, os efeitos paramagnéticos e diamagnéticos das correntes de anel são medidos facilmente e estão relacionados com compostos antiaromáticos e aromáticos.[20]
A partir da otimização dos compostos, os deslocamentos químicos absolutos (tensores de blindagem absolutos) podem ser calculados sem considerar uma referência. Para calcular os tensores (índices NICS) são considerados os valores calculados no centro dos anéis aromáticos, através de programas de mecânica quântica. Para manter a correlação entre os deslocamentos químicos, o índice NICS é computado utilizando o valor negativo do tensor. Desta forma, quanto mais negativo for o valor de NICS, maior será a aromaticidade do anel estudado.[20]
Tipos de compostos aromáticos |
A esmagadora maioria dos compostos aromáticos são compostos de carbono, mas eles não necessariamente tem de ser hidrocarbonetos.
Heterocíclicos |
Em heterocíclicos aromáticos (heteroaromáticos), um ou mais dos átomos no anel aromático é um elemento outro que não o carbono. Isso pode diminuir a aromaticidade do anel (assim como no caso do Furano), aumentando a sua reatividade. Outros exemplos incluem piridina, pirazina, imidazol, pirazol, oxazol, tiofeno, e seus análogos benzoanelados (benzimidazol, por exemplo).
Heteroaromáticos compostos por anéis de 5 membros como pirrol e furano são análogos estruturais do ânion ciclopentadienilo, pois nestes compostos o sexteto aromático é composto de duas ligações pi conjugadas e um orbital p preenchido, enquanto que os heteroaromáticos formados por anéis de 6 membros como a piridina e a borazina (B3N3H6), também conhecida como "benzeno inorgânico" são análogos estruturais do benzeno, pois neles o sexteto é formado por três ligações pi conjugadas.
A presença de diferentes heteroátomos nos diversos compostos heteroaromáticos confere a estes propriedades peculiares. Por exemplo, o pirrol apesar de ser uma amina, é consideravelmente menos básico do que o furano] e o tiofeno, um éter e um tioéter, respectivamente, comportamento distinto do observado para compostos alifáticos ou mesmo para derivados do benzeno. Isto se deve ao fato de o par eletrônico n do nitrogênio do pirrol ser parte integrante do sistema aromático, dificultando seu compartilhamento com o próton. Como os átomos de enxofre e oxigênio do tiofeno e do furano apresentam dois pares n, sendo que somente um deles é parte integrante do sistema aromático, ficando o outro disponível para compartilhamento com o próton.
Quando comparamos anéis de 5 e 6 membros contendo o mesmo heteroátomo, também se observam diferenças na estrutura e reatividade. A piridina é significativamente mais básica do que o pirrol, pois em sua estrutura o par n do nitrogênio não é parte do sistema aromático, tal como o segundo par do furano e do tiofeno, caso discutido no parágrafo anterior. Quando comparamos o furano com o pirano e o tiofeno com o tiopirano é notável que os anéis de 6 membros não apresentam caráter aromático, uma vez que não há possibilidade de conjugação entre as 2 ligações pi carbono-carbono presentes nas estruturas do tiopirano e do pirano e qualquer dos pares n do enxofre ou do oxigênio, respectivamente.
Policíclicos |
Os hidrocarbonetos aromáticos policíclicos são moléculas contendo dois ou mais anéis aromáticos simples fundidos através da partilha de dois átomos de carbono vizinhos (ver anéis aromáticos simples). Podem ser construídos a partir do Benzeno e como exemplo temos o Naftaleno, o Antraceno e o Fenantreno.[21] Moléculas aromáticas exibem maior estabilidade do que seria esperado para a estrutura de Kekulé individual. Na teoria de ligação de valência, a diferença de energia do composto real e aquela representada pela maioria das estruturas canônicas estáveis é definida como Energia de Ressonância, que é um conceito teórico que provê um índice útil para a estabilidade da molécula em relação àqueles esperados para a fórmula estrutural usualmente escrita. Compostos com maior número de anéis tendem a ter menor energia de ressonância por elétron π do que o benzeno.[21][22]
No entanto, correlacionar a estabilidade com a energia de deslocalização de Hückel dá uma correlação pobre e só foi conseguido quando Hess e Schaad usaram um Polieno como referência. A energia de ressonância de Hückel foi calculada como a diferença entre a energia total do Orbital Molecular de Hückel (HMO) e a energia da estrutura localizada.[23]
RE = Eπ(molécula conjugada) - Eπ(molécula de referência)
Hess e Schaad apresentaram um grande número de valores de REPE, abreviação para Resonance Energy Per Electron (Energia de Ressonância por elétron) para uma coleção de hidrocarbonetos benzênicos. Então, tornou-se claro que a energia varia drasticamente em cada um dos grupos dos sistemas estudados. Aplicação de HOMA, Harmonic Ocillator Model of Aromaticity (Modelo do Oscilador Harmônico para a Aromaticidade) e REC, Ring Energy Content (Conteúdo de Energia no Anel) permitiu que pesquisadores considerassem estas variações levando em conta a aromaticidade local (Quando os índices são calculados para cada anel individual).[24]
Uma outra manifestação da estabilidade aromática é a resistência a reações de adição. Por exemplo, o Ea e ΔH para a cicloadição com o etino foi calculada, e os resultados são mostrados abaixo. Os anéis internos tem maior exotermia e mais baixa Ea.[22]
| Composto | Ea | ΔH |
|---|---|---|
| Benzeno | 43.8 | 6.1 |
| Naftaleno | 36.8 | −8.7 |
| Antraceno (anéis externos) | 34.3 | −13.9 |
| Antraceno (anel central) | 29.4 | −26.2 |
| Naftaleno (anel externo) | 33.3 | −16.1 |
| Naftaceno (anel interno) | 26.8 | −32.6 |
| Pentaceno (anéis externos) | 32.7 | −17.3 |
| Pentaceno (anéis internos) | 25.5 | −35.4 |
| Pentaceno (anéis centrais) | 24.0 | −39.5 |
Uma tendência similar é observada para os índices de reações de adição Diels-Alder do antraceno, naftaleno e pentaceno, na qual três, quatro e cinco anéis, respectivamente, são linearmente fundidos.[22]
Uma tendência similar é observada para os índices de reações de adição Diels-Alder do antraceno, naftaleno e pentaceno, na qual três, quatro e cinco anéis, respectivamente, são linearmente fundidos.
O benzeno pode ser fundido de maneira angular, como no fenantreno, o criseno e o piceno, e são moléculas completamente reativas no anel central frente a reações de adição. E isso pode ser notado pelos valores de NICS (nucleus-independent chemical shift) mostrados abaixo.

Como podemos ver para o fenantreno, temos no anel central o mais baixo valor. Isso é devido à natureza localizada do anel.
Substituídos Aromáticos |
Muitos compostos químicos são anéis aromáticos com outras estruturas anexas. Exemplos incluem trinitrotolueno (TNT), ácido acetilsalicílico (aspirina), paracetamol, os nucleotídeos do DNA.
Compostos aromáticos atípicos |
Aromaticidade é também encontrada em íons como o cátion ciclopropenilo (sistema 2e), o ânion ciclopentadienilo (sistema 6e), o íon tropilium (6e) e o diânion ciclooctatetraeno (10e). Têm sido atribuídas propriedades aromáticas a outros compostos benzênicos, como a tropona. Propriedades aromáticas são testadas ao limite em uma classe de compostos chamados ciclofanos.
Um caso especial de aromaticidade é encontrado na homoaromaticidade onde uma conjugação é interrompida por um único átomo de carbono hibridizado sp³. Quando o carbono no benzeno é substituído por outros elementos, como no borabenzeno, silabenzeno, germanabenzeno, estanabenzeno, fosforina, arsabenzeno ou sais de pirilium a aromaticidade é ainda mantida.
Também ocorre aromaticidade em compostos que não são feitos de carbono. Compostos inorgânicos de anéis de seis membros análogos ao benzeno foram sintetizados. Silicazina (Si6H6) e borazina (B3N3H6) são estruturalmente análogos ao benzeno, com os átomos de carbono substituídos por outro elemento ou elementos. Na borazina, o boro e átomos de nitrogênio alternam-se ao redor do anel.
Acredita-se que existe aromaticidade metálica em certos grupos de ligas de alumínio. A aromaticidade Möbius ocorre quando um sistema cíclico de orbitais moleculares, formados a partir de orbitais atômicos pπ e preenchidos em um nível fechado por 4n (n é um inteiro) elétrons, é dada uma única meia-torção para corresponder a uma fita de Möbius. Devido poder ser uma torção canhota ou destra, os compostos aromáticos Möbius resultantes são dissimétricos ou quirais. Até agora não há nenhuma dúvida sobre a prova de que uma molécula de aromático Möbius tenha sido sintetizada.[25][26] Aromáticos com duas meias-voltas correspondente à topologias paradrômicas, primeiro sugeridas por Johann Listing, foram propostas por Henry Rzepa em 2005.[27] Nos carbômeros as ligações do anel são estendidas com grupos alcinos e alenos.
Anulenos |
Os anulenos são hidrocarbonetos monocíclicos compostos de ligações duplas conjugadas onde o número de carbonos sp2 é representado por colchetes como por exemplo [4]-anuleno e [6]-anuleno como mostra a figura 1.[28]

É uma estrutura macrocíclica que em geral apresenta boa reatividade quando ocorre a fusão de anéis benzênicos para a formação de benzoanulenos (1) ou dihidrobenzoanulenos (2). Por outro lado, não apresenta boa reatividade quando ocorre uma substituição de uma ligação dupla por um acetileno para a formação de dihidroanulenos (3).

O critério de aromaticidade também é aplicado aos anulenos. Entretanto, alcançar a planaridade torna-se uma tarefa difícil para anéis muito grandes devido aos efeitos estéricos e de torção dos ângulos das ligações. Se um anel que possua 4n+2 elétrons π é grande o suficiente para que a planaridade não cause os efeitos descritos acima, ele provavelmente adotará a conformação planar e será estabilizado devido à deslocalização eletrônica, tornando-se assim aromático. Anulenos que apresentam grandes anéis e 4n elétrons π tendem a modificar sua conformação, deixando de ser antiaromático e adotando uma estrutura não planar e não-aromática, que possui menor energia. O [6]-anuleno é uma substância aromática clássica e o [8]-anuleno que não é aromático.[28][29]
Há poucos relatos de reações de transformações químicas em anulenos na literatura, este fato se deve principalmente à sua elevada capacidade de tautomerização mesmo em condições de baixa temperatura. Como exemplo, têm-se o [10]anuleno, o menor anuleno passível de adotar diferentes conformações, como as relatadas por Castro e cols., em 2006.[30]

No [10]-anuleno, existe um grande efeito estérico entre os hidrogênios nas posições 1 e 6. A forma planar exige um ângulo de 144º entre os carbonos, o qual é muito largo para acomodar uma estrutura com carbonos sp2. O sistema prefere uma conformação não planar e não é aromático. A formação de uma ponte entre os carbonos 1 e 6 gera uma estrutura que é razoavelmente plana e aromática, onde os comprimentos das ligações variam de 1.37-1.42A.[31]

O [12]-anuleno (4n elétrons π) é uma molécula antiaromático e só é estável a temperaturas muito baixas. A formação do seu diânion estabiliza a molécula tornando-a aromática (4n+2 elétrons π).

No [18]-anuleno, a cavidade interna de sua estrutura é suficientemente grande para tornar pequenos os efeitos estéricos dos hidrogênios internos. A molécula é quase planar e mostra aromaticidade. A energia de ressonância estimada é 37 kcal/mol, que é semelhante a do benzeno.

Em geral, os anulenos também conduzem a produtos resultantes de reações de protonação, acilação, alquilação e dicátions do tipo (A+2) através de uma espécie cátion-radical, com subseqüente reação de oxidação. Estas moléculas por sua vez, podem funcionar como excelentes agentes de redução, como o caso do diânion, ciclooctatetraeno que tem sido muito usado na redução de nitro derivados do benzeno, quinonas e até de cátions, como o íon tropílium.[32]
Diversos métodos de obtenção dos anulenos ou dihidroanulenos são descritos na literatura, porém dois métodos são mais utilizados para síntese de anulenos.
O primeiro descreve o acoplamento oxidativo de um diacetileno terminal a um poliacetileno macrocíclico como intermediário chave, por rearranjo prototrópico leva a um dihidroanulenos completamente conjugado e por hidrogenação catalítica levou ao anuleno correspondente.
Como exemplo, a síntese de [18] anulenos mostra bem o método de acoplamento oxdidativo do 1,5-hexadieno (4) levando a um poliacetileno de 18 membros como intermediário (5), o rearranjo prototrópico do 1,5-hexadieno leva ao 1,7,13-tridihidro[18]anuleno (6) que é convertido através da hidrogenção catalítica no [18] anuleno (7).[29][33]

Outra rota bastante utilizada para a síntese de anulenos parte da abertura fotolítica de anéis policíclicos, porém somente o [16] anuleno foi sintetizado por essa síntese. É obtido através da fotólise do dímero do ciclooctatetraeno em éter a 0°.[34]
Efeito Mills-Nixon |
Em 1930 Willians Hobson Mills e Ivor Gray Nixon publicaram um artigo no qual propuseram nova abordagem sobre aromaticidade.
Segundo eles, de acordo com a estrutura proposta por Kekulé, um anel aromático não apresentaria suas nuvens eletrônicas externas direcionadas a partir de seu núcleo, mas sim de fora do anel formando um ângulo que seria variável de acordo com as configurações dos átomos de carbono de dupla ligação, ou seja, os ângulos e comprimentos das ligações eram variáveis de acordo com o tipo de ligação, se dupla ou simples.
A determinação dos ângulos era um desafio, pois não era percebida diferença considerável entre os átomos de carbono presentes na molécula, já que eles estavam em constante mudança, onde duplas ligações se alternavam quando era formada outra entre dois átomos de carbono.
De acordo com sua teoria, Mills e Nixon propuseram que sintetizando anéis benzênicos fundidos a anéis de 5 ou de 6 membros, seria possível chegar a um composto no qual a ligação entre os anéis seria feita através de uma ligação simples, que de acordo com os pesquisadores, é mais estável e é a forma preponderante no equilíbrio quando no estado líquido ou em solução, pois a forma onde a ligação entre os anéis é dupla, há maior tensão intramolecular.
Como não havia nenhum método para confirmar a distribuição das ligações duplas no anel, para provar suas teorias, foram realizados experimentos diversos com indanos e anéis benzênicos fundidos e baseados em estereoquímica dos produtos obtidos, mostraram que havia mudança na orientação de reações de síntese com ciclos rígidos ligados diretamente a anéis aromáticos.[35]
Embora alguns pesquisadores afirmem que o efeito Mills-Nixon seja real e que pode ser provado experimentalmente, outros sugerem que este vem de artifícios teóricos de aproximação e que cálculos mais aprofundados demonstraram que o efeito não é real.
Em 1986, Dierck e Vallhardt publicaram a síntese e a caracterização do composto triangular[4]fenileno, representado abaixo, que foi o primeiro ciclohexatrieno sintetizado e que possui um anel central com suas ligações completamente localizadas enquanto os outros anéis benzênicos apresentam suas ligações completamente deslocalizadas. A deslocalização das ligações do anel central poderiam causar um caráter antiaromático aos três ciclobutadienos do composto, assim, a perda da aromaticidade é energeticamente favorecida uma vez que não há a formação dos três anéis antiaromáticos. Porém, esses anéis de quatro membros formam pequenos ângulos com o anel benzênico central (Θ 88,3º), sendo o efeito Mills-Nixon responsável pela localização das ligações observadas.[36]
O efeito Mills-Nixon pode ser explicado por três diferentes formas. Pela análise do sistema π, no qual os anéis fundidos ao anel central possuem 4n+2 elétrons e o sistema pode exibir ligações exocíclicas mais longas no neste que se comparados as ligações endocíclicas. A segunda forma é pela localização da ligação induzida pela tensão do anel, uma vez que o efeito tensional causa a reibridização nos átomos mais tensionados. E a última forma é pela curva formada pela ligação envolvida, a chamada ligação tipo banana. Neste caso, o ΔR (ΔR= A sin² α + B) representa a diferença entre a ligação C-C exociclica e a endocíclica no anel benzênico ciclizado com o anel fundido ao anel benzênico central, descrevendo o efeito da tensão do anel na geometria do anel benênico central.[37] Porém, quando o sistema é mais flexível, este introduz um fator adicional para compensar a tensão do anel (a formação das ligações do tipo banana) para diminuir a alternância entre as ligações na molécula e assim, aumentar a deslocalização das ligações.
No composto onde os anéis de quatro membros não dividem seus elétrons como os elétrons do anel benzênico os efeitos de aromaticidade-antiaromaticidade são mais pronunciados. Entretanto, os ângulos formados pelos anéis de quatro membros são menores e dessa forma, a tensão do anel é mais pronunciada.
Para analisar a influência da tensão do anel isoladamente, Amnon Stager investigou um sistema que não continha elétrons π os substituintes do anel benzênico, e concluiu que a tensão do anel pode ser suficiente para causar a fixação das ligações do benzeno, ou seja, a densidade eletrônica aumenta com a diminuição do comprimento da ligação e vice-versa.
Muitas vezes para compensar a tensão angular entre as ligações há a formação das ligações do tipo banana, e nesses casos o efeito Mills-Nixon não é observado.
Assim, para observar o efeito Mills-Nixon, deve-se utilizar carbonos sp² nas posições tensionadas do sistema e estes não devem participar do sistema conjugado π.
Aromaticidade de Superátomos |
Os superátomos são constituídos por um aglomerado estável ou metaestável de átomos, chamados de cluster, que podem mimetizar o comportamento químico de átomos elementares.[38]
Uns dos caminhos possíveis para a química é a utilização destes clusters estáveis, inserindo-os na síntese de materiais em nanoescala, que podem ser ajustados para criar as propriedades desejadas.[39]
De acordo com os cientistas a nova Tabela Periódica seria algo em terceira dimensão, segundo Schmidt-Ott, os superátomos identificados possuem propriedades químicas semelhantes às dos elementos da Tabela Periódica, pois suas camadas externas são praticamente as mesmas. Porém, a descoberta de superátomos com uma camada externa diferente, irá proporcionar um conjunto de propriedades completamente novas.[40]
Foram descritos o comportamento dos clusters de Al13 e Al14 através da análise das propriedades químicas, da estrutura eletrônica e de sua geometria, formando compostos químicos com átomos de iodo. Observou-se que um cluster com 13 átomos de alumínio era capaz de mimetizar o comportamento químico do iodo, já com 14 átomos de alumínio se comportava como um metal alcalino- terroso.[39]
Uma das metas de Schmidt-Ott é encontrar clusters com propriedades ópticas, magnéticas ou elétricas totalmente novas, que sejam suficientemente estáveis para criar cristais ou outras formas sólidas.[40]
Khanna e colaboradores desenvolveram um trabalho sobre a síntese de uma molécula inorgânica cíclica incomum de arsênio e telúrio, As2Te22-, onde foram encontradas características magnéticas e aromáticas.[41]
Os resultados obtidos por Khanna publicados pela revista Journal of the American Chemical Society, sugerem que os elementos tradicionalmente não magnéticos podem tornar-se magnético. Este fato amplia a compreensão da aromaticidade e mostra que ela pode ocorrer em materiais inesperados.[42]
Para a química orgânica é normal encontrar sistemas aromáticos estáveis com 4n + 2 elétrons π, contudo já são conhecidas moléculas aromáticas não-orgânicas.[41]
Os critérios definidos por Kekulé, foram propostos para discriminar a aromaticidade em sistemas orgânicos, esta aromaticidade é alcançada pelo envolvimento de elétrons π, embora estas regras se apliquem em muitos casos de aromaticidade e/ou antiaromaticidade σ, e ainda a aromaticidade e/ou antiaromaticidade em sistemas metal-aromático, especialmente os que exibem multipla-aromaticidade que é rara em compostos orgânicos e inorgânicos, no entanto parece ser comum em sistemas metal-aromáticos.
Não se pode esperar que a aromaticidade/antiaromaticidade em sistemas metal-aromáticos se manifestem da mesma maneira que em sistemas orgânicos, no entanto muitas das propriedades químicas destes compostos só se explicam se empregarmos o conceito de aromaticidade. Um maneira de estudarmos aromaticidade e antiaromaticidade em sistemas metálicos é a construção de clusters metálicos.
Clusters metálicos como o MAl4- , são um bom ponto de partida para o estudo de aromaticidade em sistemas metal-aromático, isto por que estão livre influência de substituintes, do reticulo cristalino e do solvente.
O clusters do tipo MAl4- são muito semelhantes entre si , formam pirâmides de base quadrada, onde as unidades Al42- formam a base, independente do metal envolvido. Mesmo com pequenas mudanças na unidade Al42-, para a formação da espécies MAl4- a integridade da estrutura piramidal é preservada, assim como os compostos sanduíches com benzeno do tipo M(C6H6).
O diânion Al42- é triplamente aromático e isto se deve aos três tipos de orbitais moleculares formados pelos orbitais atômicos 3p do átomo de alumínio : pπ (orbitais π perpendiculares a base da quadrada da estrutura piramidal), pσ-r (orbitais orientados radialmente em direção ao centro da base da quadrada da estrutura piramidal), pσ-t (orbitais orientados tangencialmente ao redor da base da quadrada da estrutura piramidal). Estes orbitais moleculares conferem uma forma quadrada perfeita e consequentemente a tripla aromaticidade natural.
As propriedades aromáticas de espécies Al42- também são estudadas através da função de localização de elétron (FLE). A região do espaço com maior valor de FLE, corresponde a região de maior probabilidade de encontrar um par de elétron. Os resultados demonstram que no caso do diânion Al42- a deslocalização dos elétrons σ são mais importantes do que a deslocalização dos elétrons π . De fato Al42- é uma das espécies mais aromáticas com base nos resultados da análise de bifurcação de FLE.[43]
Aromaticidade por Orbitais Moleculares |
A teoria de orbitais moleculares (OM) é uma teoria bem aceita pela comunidade química para entender a aromaticidade por conseguir explicar a estabilidade diferenciada de certas moléculas e além disso conseguir prever a instabilidade de algumas moléculas cíclicas.
Tal teoria descreve os orbitais σ e π como sendo ortonormais. Dessa forma, a abordagem para os elétrons σ e π é diferente. A molécula é analisada como um todo, porém o esqueleto é "montado" pelos elétrons σ e este por sua vez é "vestido" com os elétrons π.
A expressão mais simples de entrecruzamento entre OM e aromaticidade é a regra de Huckel. Esta regra prevê que os hidrocarbonetos cíclicos, planares e completamente conjugados serão aromáticos se possuírem 4n+2 elétrons π. Sendo n qualquer número inteiro.
Exemplos dessa regra podem ser verificados na figura 1, onde temos estruturas classificadas como aromáticas quando seus elétrons estão dispostos abaixo da linha de base (geralmente é no nível de energia dos orbitais p isolados), diminuindo sua energia; não aromáticas (aquelas que não apresentam diferenças de energia em relação ao seu alqueno similar); e as estruturas antiaromáticas (as que possuem orbitais antiligantes ocupados), que apresentam energia molecular muito alta e tendo geralmente um tempo de vida muito curto.[44]

Figura 1: Energias de Huckel para os orbitais de alguns anéis conjugados.
Orbital Molecular da Estrutura do Benzeno |
No benzeno, os átomos de carbono apresentam ângulo de 120° sugerindo uma hibridização sp2. Com isso, a representação ideal de um anel de seis membros, plano, com cada átomo de carbono sp2 possuindo um orbital p disponível para superposição com os orbitais p dos carbonos vizinhos, como mostra a figura 2.[45]
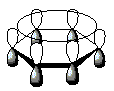
Figura 2: Representação estabilizada dos orbitais p do anel benzênico.
Como os seis orbitais atômicos p combinam-se para formar seis orbitais moleculares π, três dos orbitais moleculares tem energias mais baixas que a de um orbital p isolado (orbitais moleculares ligantes), os outros três orbitais moleculares tem energias mais altas que a de um orbital p isolado (orbitais moleculares antiligantes), como mostrado na figura 3.

Figura 3: Orbitais atômicos combinam-se dando origem a orbitais moleculares.
O benzeno em sua estrutura eletrônica do estado fundamental apresenta seis elétrons π nos orbitais moleculares π, de menor energia. O orbital molecular π de mais baixa energia tem superposição de orbitais p com os mesmos sinais matemáticos de fase em torno de toda face de cima e de baixo do anel (não há plano nodal). Cada um dos dois próximos orbitais, de mais alta energia, apresentam um plano nodal e por isso estes possuem a mesma energia (orbitais degenerados) . Estes três orbitais formam o conjunto de orbitais moleculares ligantes do benzeno. O próximo conjunto de orbitais de mais alta energia (π*) apresentam dois planos nodais em cada um dos orbitais, e o de mais alta energia possui três planos nodais. Esses três são os orbitais moleculares antiligantes do benzeno e encontram-se desocupados no estado fundamental (figura 4).[46]
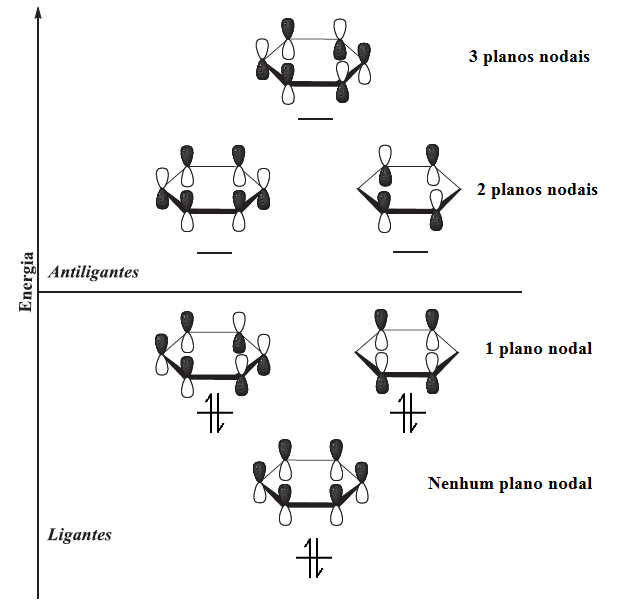
Figura 4: Representação gráfica dos orbitais moleculares do benzeno.
Sabe-se da enorme diferença de estabilidade entre o ciclobutadieno e o benzeno. Isso se deve basicamente ao ciclubutadieno apresentar dois elétrons desemparelhados por conta da degeneração de dois orbitais em seu diagrama. Esse dois elétrons desemparelhados não contribuem para a estabilização da molécula e por serem de alta energia geralmente são usados para fazer reações químicas. Porém este composto pode fugir da antiaromaticidade prevista por Huckel se contorcendo e assumindo assim uma forma retangular e se tornado assim uma molécula não aromática com energia próxima a do butadieno.
Compostos ditos aromáticos também são caracterizados por apresentarem um grande salto de energia entre seus orbitais HOMO e LUMO.
Para o benzeno, o calculo de Huckel gera o valor de 6α + 8β, menor 2β que a energia calculada para três duplas ligações isoladas, que seria de 6α + 6β. Essa diferença de energia é chamada de energia de deslocalização ou energia de ressonância.
Há uma segunda aproximação também usada na regra de Huckel. Este método apresenta o melhor desempenho e nele se trata a energia de ressonância não por parâmetros α e β, mas sim de maneira quatitativa. A abordagem termoquímica prevê uma energia de ressonância de aproximadamente 36Kcal/mol do benzeno em relação ao 1,3,5 hexatrieno(figura 5).

Figura 5: Diferença de energia entre o benzeno e o cicloexatrieno.
Espécies Aromáticas Carregadas |
A aromaticidade em anéis com carga pode ser avaliada pelos critérios de aromaticidade dos anuleis neutros. Assim os níveis de energia HMO e o rol de Huckel podem ser critérios energéticos e eletrônicos aplicáveis a cátions e ânions que tem uma estrutura completamente planar conjugada, considerados como compostos aromáticos (Figura 1).[47]

Figura 1. Cátions e anions que seguem a regra de Huckel (4n+2 elétrons π). A) cátion ciclopropenílico, B) dicátion ciclobutadieno, C) diânion ciclobutadieno, D) ânion ciclopentadieno, E) cicloheptatrieno (íon tropílio), F) dicátion ciclooctatrieno, G) diânion ciclooctatrieno.
Os cátions ciclopropenílico (A) e cicloheptatrieno (íon tropilio) (E) e o ânion diclopentadieno (D) são os exemplos mais populares deste tipo de compostos aromáticos em anéis com carga. O cátion ciclopropenílico é o menor membro do sistema aromático de Huckel, contendo 2π elétrons deslocalizados em três orbitais 2p (Figura 2).
Este tipo de cátion tem uma considerável estabilidade termodinâmica, que pode ser medida através de um parâmetro empírico, o valor de pKR+. Este valor corresponde ao pH da solução quando o carbocátion é 50% neutralizado. Cátions ciclopropenílico com vários substituintes apresentam rangos de -0.67 a 10.0 enquanto o cátion ciclopropenium não substituído apresenta o valor de -7.4, o qual é um valor intermédio entre íons altamente estáveis como o cátion trifenilmetil. Derivado deste tipo de cátion há sido um importante aspecto desde uma perspectiva teórica e importantes aplicações[48]
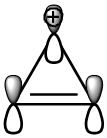
Figura 2. Orbitais 2p do cátion ciclopropenílico. Sistema aromático de Huckel (4n+2) elétrons π.
Algumas espécies insaturadas não são aromáticas, mas possuem algumas características estruturais de anéis aromáticos como, serem cíclicos e planares. Porem não possuem uma sequência ininterrupta de orbitais p ligantes. No entanto algumas dessas espécies quando passam para sua forma ionizada (reibridização) ganham aromaticidade seguindo a regra de Huckel (4n+2 elétrons π) um exemplo é o ciclopentadieno (figura 6) que possui uma grande facilidade de perda de um próton (H ácido) devido a grande estabilidade de sua base conjugada (uma espécie aromática aniônica.

Figura 6: Ionização do ciclopentadieno.
O diagrama de orbitais dessa espécie (figura 7) mostra que, com seis elétrons, os três orbitais ligantes estão ocupados e os dois antiligantes vazios, indicando assim a aromaticidade do composto.

Figura 7: Diagrama de orbitais do ciclopentadieno.
O mesmo vale para espécies como o cicloheptatrieno (figura 8), que se encaixa na regra de Huckel quando se encontra em sua forma catiônica (cátion Tropílio).
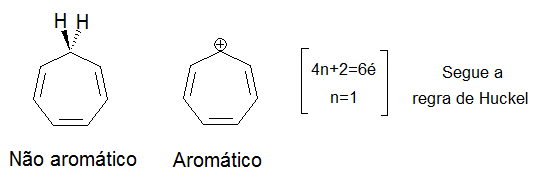
Figura 8: Cicloheptatrieno e seu análogo carregado, o cátion tropílio.
O diagrama de orbitais do cátion tropílio (figura 9) mostra que, com seis elétrons, os três orbitais ligantes estão ocupados e os quatro antiligantes vazios, confirmando assim a aromaticidade do composto.

Figura 9: Diagrama de orbitais do cátion tropílio.
Aromaticidade pela teoria de Ligação de Valência |
Em 1916 Lewis descreveu uma ligação química como sendo dois elétrons compartilhados por dois átomos.[49] Logo mais tarde, em 1927, Heitler e London publicaram o tratamento quantum-mecânico da molécula de hidrogênio seguindo os preceitos de Lewis, o que marcou o início da teoria da ligação de valência.[50] Em 1933, Pauling e Whelang publicaram uma teoria de ligação de valência simplificada, que foi sendo aprimorada por outros grupos posteriormente.[51] O conceito de valência está relacionado com o número de elétrons disponível para cada átomo, onde o máximo de estabilidade significa alcançar o número de quatro pares de elétrons seguindo a regra do octeto.
A teoria de ligação de valência é simples para moléculas menores, no entanto, no caso de moléculas maiores a representação dá-se pelo o maior número de estruturas de Lewis possíveis para tal composto. A teoria de ligação de valência assume que a representação correta é um híbrido destas formas canônicas. Para moléculas maiores, isso torna-se um problema devido ao número de formas canônicas possíveis. Para tentar solucionar estes problemas, a teoria de ligação de valência adotou conceitos como hibridização e ressonância.[52]
Embora a teoria de ligação de valência forneça uma ideia muito mais clara do que é uma ligação química estruturas conjugadas são muito melhor descritas através da teoria do orbital molecular. O conceito de ressonância adotado pela teoria de ligação de valência auxilia na descrição de sistemas conjugados. A teoria de ressonância é uma extensão da teoria de ligação de valência que reconhece que para algumas moléculas mais de uma estrutura de Lewis pode ser escrita. Este conceito é interessante para caracterizar a deslocalização eletrônica característica de sistemas conjugados.
A teoria de valência proposta por Pauling e Whelang falha em explicar antiaromaticidade e não consegue tratar efeitos magnéticos como exaltação de susceptibilidades diamagnéticas. Apesar de diversas tentativas em aprimorar a teoria de valência clássica [53][54][55] somente em 1970 Mulder e Oosterhoff encontraram uma maneira de resolver estes problemas.[56] Eles descobriram que no caso de sistemas monocíclicos, termos específicos na equação de energia da teoria de ligação de valência descrevem o efeitos desestabilizante de anéis com 4n elétrons π e efeitos estabilizantes em anéis com 4n + 2 elétrons π. Este evento é conhecido como permutação em anel. Através do conceito de permutação de anel tanto a antiaromaticidade como os efeitos magnéticos podem ser explicados.[57]
Tantardini e colaboradores publicaram cálculos ab initio seguindo a teoria de ligação de valência para o benzeno. Eles concluíram que a estrutura de Kekulé é a mais estável e mais ligante, e ainda, o seu número de ocupação na função de onda do estado fundalmental é o mais alto.[58] Mais recentemente, em 1995, Li e Jiang preveram uma série de parâmetros como comprimento de ligação, reatividade e aromaticidade local de alguns hidrocarbonetos benzenóides utilizando o método Lanczos em um contexto de teoria de ligação de valência. Os resultados obtidos estão de acordo com os dados experimentais e com dados obtidos através da teoria do orbital molecular.[59]
O modelo VB para o benzeno leva em consideração as duas estruturas de Kekulé e as três estruturas de Dewar. A função de onda pode ser descrita conforme abaixo onde c1 é o coeficiente e ϴ1 a função de onda da estrutura 1.

Como dito já anteriormente, as estruturas de Kekulé são mais importantes que as estruturas de Dewar e isto é expresso nos valores dos coeficientes. A função de onda ψ pode ser avaliada em termos da integral coulombica Q, que corresponde aproximadamente ao Hrr no tratamento pela teoria do orbital molecular, e também pela integral de troca J, que representa a troca de dois elétrons entre um par de átomos de carbono. A energia de elétrons π do benzeno pode ser avaliada nestes termos, uma determinante secular 5 X 5 é formada, as raízes são os valores E = Q + 2,16J, Q — 4,16J, Q — 2J e Q — 2J. A menor energia tem o valor E = Q + 2,16J, enquanto que a energia de uma única estrutura de Kekulé é Q + 1,5J. A diferença em energia, 0,66J representa o aumento de estabilidade do benzeno em relação a uma única estrutura de Kekulé.
Aromaticidade pela teoria de Átomos em Moléculas |
R.F.W. Bader e colaboradores, foram uns dos pioneiros a descreverem a distribuição eletrônica de um especifico átomo em uma molécula, chamado método de átomos em moléculas(AIM). As bases dessa teoria deriva de mecanismos quanticos e princípios físicos.A função de onda ᴪ, com seu complexo conjugado ᴪ* fornecem a probabilidade de se encontrar a partícula em um determinado ponto. Se temos N elétrons em uma molécula, P(r) será a densidade de probabilidade de se encontrar um elétron por unidade de volume. Pela integração de P(r) obtemos a densidade eletrônica designada por ρ(r), onde a mesma integrada sobre todo o espaço é igual a N. A densidade eletrônica é mostrada através de uma série de contornos, onde o “caminho da ligação” (bond paths) é o caminho de máxima densidade eletrônica entre dois átomos. O ponto crítico é o ponto no caminho da ligação onde a densidade eletrônica é maxima ou mínima com respeito a deslocalização em qualquer direção. O ponto crítico da ligação é definido pela equação:
∇¯ρ(r)⋅N¯(r)=0{displaystyle {overline {nabla }}rho (mathbf {r} )cdot {overline {N}}(mathbf {r} )=0}

O ponto crítico é o ponto em que o campo vetor gradiente de densidade de carga é zero, ou seja, um limite máximo ou mínimo ao longo de N. Esta condição aplicada a outros caminhos entre dois átomos define uma superfície única que pode representar o limite dos átomos dentro da molécula. A densidade de elétrons dentro destas fronteiras, em seguida, dá a carga atômica. A combinação de contornos densidade de elétrons,caminhos de ligação e pontos críticos define o gráfico molecular. Esta análise pode ser aplicada a densidade de elétrons calculada por métodos ou MO ou DFT. Para molécula simples, como H2, o caminho de ligação é uma linha reta entre os núcleos. Caminhos de ligação geralmente não são lineares em moléculas mais complexas. Eles são fortemente curvado em moléculas tensas, tais como aqueles contendo anéis pequenos.

A teoria de A e M também vem sendo aplicada para o estudo de aromaticidade, onde foi encontrada uma densidade de deslocalização π em hidrocarbonetos cíclicos insaturados do que em hidrocarbonetos acíclicos.

Foi encontrada uma correlação entre a densidade de carga no ponto de ligação crítica e comprimentos de ligação, onde ligação mais curta tinha maior ρ(r) e melhor elipticidade, onde esta fornece uma medida quantitativa do caráter π da ligação C=C. Estudos feitos em vários anéis, indicam que o número de elétrons compartilhado por átomos adjacentes são 0,99, 1,39 e 1,89,respectivamente, para benzeno, etano e eteno, onde os números que refletem a densidade de elétrons adicionais relativos às ligações múltiplas. Esses valores também foram calculados para vários sistemas de anéis fundidos, como mostrado abaixo. A elipticidade também foi calculado e é dado com as estruturas (números em itálico). Elipticidade aumenta com a ordem de ligação,refletindo o acúmulo de densidade eletrônica.

Estes valores revelam também a reatividade entre os sistemas de anéis. As maiores ordens de ligação são encontrados na ligação 1,2 naftaleno e em antraceno. Para fenantreno, a ordem de ligação é maior entre as posições 9 e 10 no anel central. Podemos observar na figura abaixo a densidade eletrônica nos diferentes anéis:

A imagem vai de encontro com a ideia de que a melhor estrutura para qualquer anel policíclico é aquele com o número máximo anéis de benzeno como anéis. Segundo este conceito os dois anéis em naftaleno são idênticos, mas menos aromático do que o anel de benzeno. Os anéis externos em fenantreno são mais aromáticos que o anel central, enquanto o anel central é mais aromático em antraceno
Antiaromaticidade |
Em alguns tipos de sistemas cíclicos contendo 4n elétrons π, a deslocalização destes elétrons leva a uma forte instabilidade do composto, em contraste a estabilização característica da aromaticidade. Nestes casos, o termo antiaromático é empregado para descrever estes sistemas [60]. Como exemplos de anéis antiaromáticos podemos citar o cátion ciclopentadienil, ânion ciclopropenil, cátion indenil, cátion fluorenil e espécies neutras como o ciclobutadieno.
Cátion ciclopentadienil |
O interesse por cátions ciclopentadienil começou em 1925 quando Ziegler e Schenell na intenção de sintetizar o radical estável pentafenilciclopentadienil (a partir do pentafenilciclopentadienol em meio ácido sulfúrico concentrado) obtiveram um composto com cor violeta intensa , o cátion pentafenilciclopentadienil. No entanto, as reações com ácido clorídrico e ácido bromídrico resultaram nos haletos ciclopentadienil [61].

Cálculos seguindo a teoria do orbital molecular de Hückel demonstraram que o cátion ciclopentadienil deveria ser um tripleto no estado fundamental [62]. Mais tarde, o cátion pentafenilciclopentadienil foi caracterizado por espectroscopia de ultravioleta e ressonância de spin eletrônico (ESR) e demonstrou ser um tripleto em estado excitado de baixa energia [63][64]. O cátion ciclopentadienil é um dos melhores exemplos de carbocátion instável. Segundo a teoria de Hückel as energias de deslocalização do cátion, radical e ânion ciclopentadienil são respectivamente 1,24β, 1,85β e 2,47β [62]. O cátion ciclopentadienil mais tarde foi sintetizado e a espectroscopia ESR demonstrou que ele é um tripleto em estado fundamental assim como foi previsto pela teoria de Hückel [65].
Cátions indenil e fluorenil |
São derivados benzanelados do cátion ciclopentadienil que demonstram antiaromaticidade atenuada. Embora o cátion indenil seja inambiguamente antiaromático existem controvérsias sobre a antiaromaticidade do fluorenil [66].

Dicátions Bisfluorenila |
O dicátion bisfluorenila e seus derivados têm sido gerados experimentalmente e estudados. Em experimentos de RMN do dicátion observam-se sinais em δ 5,07 ppm e δ 5,87 ppm, com um centro de gravidade em δ 5,41 ppm. Já o precursor neutro apresenta deslocamentos de campo mais alto apresentando centro de gravidade em δ 7,75 ppm. Estes resultados indicam que o sistema é substancialmente paratrópico e que supostamente temos uma espécie antiaromática [67].

Novas Teorias |
A aromaticidade é também analisada pela teoria AIM (átomos em moléculas). A teoria AIM permite obter informações sobre uma molécula inteira a partir de um fragmento da molécula e é baseada na análise topográfica da função de distribuição da densidade eletrônica, que pode ser obtida por métodos quânticos ou baseado em informações de difração de raio-X de alta resolução.[68]
A aromaticidade é usualmente avaliada por critérios energéticos (entalpia de hidrogenação, energia de ressonância de Dewar), magnéticos (NICS - Nucleus independent chemical shift) e estruturais (HOMA- Oscilator model of aromaticity).[69]
O conceito de aromaticidade é mais facilmente compreendido quando é discutida por localização e deslocalização eletrônica. Infelizmente localização e deslocalização eletrônica não podem ser experimentalmente medidas. Assim são usados métodos teóricos para a quantificação da distribuição eletrônica em uma molécula.
A partir do modelo AIM foram propostos três (3) índices de aromaticidade:
DI (delocalization index) |
Índice mais utilizado na quantificação de aromaticidade. Fornece a medida do número de elétrons compartilhados por dois átomos ou bacias. Quanto maior o valor de DI, mais disperso os elétrons estão na molécula. Vide DI do hexatrieno, molécula acíclica com ligações duplas conjugadas, e do benzeno.[70]
| Tipo de ligação | Valores de DI |
|---|---|
| Ligação C-C do etano | 1,0 |
| Ligação C-C do benzeno | 1,39 |
| Ligação C-C do hexatrieno | 1,14 |
| Ligação C=C do hexatrieno | 1,74 |
| Ligação C-C do Íon tropílico | 1,365 |
D3BIA (density, degeneracy, delocalization-based Index of Aromaticity) |
Baseado na densidade no anel, no DI e na degeneração que é proporcional à distribuição uniforme dos elétrons entre os átomos do anel. Segundo esse índice, quanto maior o tamanho do anel e o número de heteroátomos, menos aromática é a molécula.[70]
Teoria isoeletrônica complexa (Cplex-isoelectronic theory):[71] |
É a primeira teoria de sucesso, que não se baseia na Mecânica Quântica, proposta em 2007. Parte de três princípios: ADEP (antiperiplanar dynamics of isoelectron pairs), SDEP (synperiplanar dynamics of single isoelectrons) e SDSE (synperiplanar dynamics of isoelectrons pairs). A hipótese ADEP refere-se ao movimento de isoelétrons de modo antiperiplanar em relação ao plano da molécula. O princípio SDEP refere-se ao movimento de isoelétrons desemparelhados de modo sinperiplanar em relação ao plano da molécula. Já a suposição SDSE refere-se ao movimento de isoelétrons de modo sinperiplanar em relação ao plano da molécula.
Esta teoria é consistente com a Química Hadrônica de Santilli, que supõe que os elétrons de uma ligação química sobrepõem de modo a criar um estado de quase partícula (isoelectronium). Está de acordo também com a teoria de Robson e Ingold. É uma teoria qualitativa que faz conexões lógicas entre sistemas conhecidos e desconhecidos em nível químico e é aplicada à química pericíclica e aromaticidade.
A aromaticidade é definida como um processo contínuo ADEP que pode ser iniciado com a mesma probabilidade acima ou abaixo do plano da molécula e com densidade eletrônica seletiva sin facial final (syn FSED — syn final facial selectivity of electron density), ou seja, a face sobre a qual ocorre uma diminuição da densidade eletrônica devido ao processo ADEP corresponde à mesma face onde ocorre aumento da densidade eletrônica como um circuito espiroidal, o que gera um caráter de dupla ligação.
Evidências experimentais podem ser observadas através da aplicação de um campo magnético externo o que gera uma corrente diamagnética no anel. As linhas do campo magnético e o efeito anisotrópico são similares àqueles produzidos por uma corrente elétrica em um circuito metálico quando submetido a um campo magnético.
Baseado nesta teoria, anéis neutros com número ímpar de duplas ligações (4n +2 isoeletrons) e sistemas com número par (4n), com exceção do ciclobutadieno, com processo SDEP, são preditos aromáticos.
Teoria do funcional da densidade |
O uso da Teoria do Funcional da Densidade (DFT, do inglês Density Functional Theory) tem se mostrado como uma importante ferramenta no estudo e previsão de aromaticidades de compostos, juntamente com outros cálculos computacionais oriundos da mecânica quântica, corroborados com resultados experimentas de RMN de 1H e 13C.
Na espectroscopia de Ressonância Magnética Nuclear de hidrogênio e carbono (RMN de 1H e 13C), a validação do deslocamento químico dos sinais é indispensável para caracterização estrutural de compostos. Neste contexto o conceito de eletronegatividade é útil na previsão dos deslocamentos químicos. No caso do TMS (tetrametilsilano) a densidade eletrônica ao redor dos prótons é alta (o silício é eletropositivo em relação ao carbono), logo os prótons devem estar blindados e o sinal correspondente aparecerá em valor alto de campo, sendo usado como substância padrão para análises centrado em δ 0,0 ppm. Já no caso de moléculas onde existem átomos eletronegativos ligados ao carbono, os prótons estarão desblindados e os picos correspondentes aparecerão em valores altos de campo.[72]
No entanto o conceito de eletronegatividade não é suficiente para justificar o deslocamento de certos grupos, quando observamos os sinais relativos aos prótons aromáticos, temos um efeito adicional a eletronegatividade, uma vez que, o sinal dos prótons se encontram por volta de 7ppm em RMN 1H , e por volta de 130ppm em RMN de 13C.Neste caso, deve ser observado o anisotropismo (blindagem diamagnética): os elétrons de valência dos prótons são forçados a circular produzindo uma corrente diamagnética que gera um campo magnético induzido de direção oposta ao campo magnético aplicado.[73] No que diz respeito ao benzeno , quando é induzido um campo elétrico, os elétrons π do anel aromático circulam ao redor do anel (corrente de anel), desblindando os prótons do anel. No entanto, o centro do anel está blindado, uma vez que, se encontra em sentido oposto ao campo magnético da periferia.
Metodologias como DFT e GIAO (gauge-including atomic orbital), foram utilizadas para avaliar aromaticidade correlacionando o forte efeito anisotrópico positivo (blindagem) em prótons de metanos orientados perpendicularmente ao anel aromático de determinados compostos, em comparação a compostos não aromáticos. Para ilustrar esse efeito foram escolhidos compostos aromáticos segundo a teoria de Hückel, evidenciando componentes magnéticos de aromaticidade, que mostraram correlação entre efeito anisotrópico e aromaticidade.[74]
Ver também |
- Hidrocarbonetos aromáticos
- Aminas aromáticas
- Anel aromático simples
- Homoaromaticidade
- Aromaticidade Möbius
- Aromaticidade metálica
Referências
↑ P. v. R. Schleyer, "Aromaticity (Editorial)", Chemical Reviews, 2001, 101, 1115-1118. DOI: 10.1021/cr0103221 Abstract.
↑ A. T. Balaban, P. v. R. Schleyer and H. S. Rzepa, "Crocker, Not Armit and Robinson, Begat the Six Aromatic Electrons", Chemical Reviews, 2005, 105, 3436-3447. DOI: 10.1021/cr0103221 Abstract.
↑ P. v. R. Schleyer, "Introduction: Delocalization-π and σ (Editorial)", Chemical Reviews, 2005, 105, 3433-3435. DOI: 10.1021/cr030095y Abstract.
↑ Krygowski, R.M.; Cyranski, M.K.; Czarnocki, Z.; Hafelinger, G.; Katritzky, A.R.; Tetrahedron 2000, 56, 1783
↑ Kekulé, F.A.; Bull. Soc. Chim. Fr. 1865, 3, 98
↑ Chen, Z.; Wannere, C.S.; Corminboeuf, C.; Puchta, R.; Shleyer, P.V.R.; Chem. Rev. 2005, 105, 3842
↑ Evans, M.G.; Polanyi, M.; Trans Faraday Soc. 1938, 11, 24
↑ Kruszewski, J.; Krygowski, T.M., Tetrahedron Lett., 3839, 1972
↑ Kruszewski, R.M. J. Chem. Compt. Sci. 1993, 33, 70
↑ Minkin, V.I.; Glukhovtsev, M.N.; Simkin, B.Y.; Aromaticity and Aromaticity. Electronic and Structural Aspect; J. Wiley: New York, 1994
↑ Pauling, L.; Sherman, J. J. Chem. Phys. 1933, 1, 606
↑ Kistiakowski, B.; Ruhoffm, J.R.; Smith, H.A.; Vaughan, W.E.J. Am. Chem. Soc. 1936, 58, 1657
↑ Haddon, R.C.; Fukunaga, T. Tetrahedron Lett. 1980, 21, 1191
↑ George, P.; Trachtman, M.; Bock, C.W.; Brett, A.M.; J. Chem. Soc. Perkin Trans. 2, 1976, 1222
↑ George, P.; Trachtman, M.; Bock, C.W.; Brett, A.M.. Theor. Chim. Acta 1975, 38, 121
↑ Pauling, L.; J. CHem. Phys. 1936, 4, 673
↑ Julg, A.; François, P. Theor. Chim. Acta 1967, 7, 249
↑ Dauben, H.J.; Wilson, J.D.; Laity, J.L. J. Am. Chem. Soc. 1968, 90, 811
↑ Pascal, P. Revue Generale des Sciences Pures et Apppliquess 1923, 34, 388
↑ ab Schleyer, P.V.R.; Maerker, C.; Dransfeld, A.; Jiao, H.; Hommes, N.J.R.E.; J. Am. Chem. Soc. 1996, 118, 6317
↑ ab Randíc, Milan. Aromaticity of Polycyclic Conjugated Hydrocarbons. Chem. Rev., 2003, Vol. 103, No. 9.
↑ abc CAREY, Francis A.. Advanced organic chemistry: pt. A. Structure and mechanism. 5. ed. New York: Springer, 2007.
↑ Moyano, A; Paniagua, J. C. . Localized molecular orbitals of acyclic polyenes as a basis for a new approach to resonance energies. J. Org. Chem., 1986, 51 (12), pp 2250–2257
↑ Krygowski, T. M.; Cyrański, M. K. Structural Aspects of Aromaticity. Chem. Rev., 2001, 101 (5), pp 1385–1420 DOI: 10.1021/cr990326u
↑ Synthesis of a Möbius aromatic hydrocarbon D. Ajami, O. Oeckler, A. Simon, R. Herges, Nature; 2003; 426 pp 819.
↑ Investigation of a Putative Möbius Aromatic Hydrocarbon. The Effect of Benzannelation on Möbius [4 n]Annulene Aromaticity Claire Castro, Zhongfang Chen, Chaitanya S. Wannere, Haijun Jiao, William L. Karney, Michael Mauksch, Ralph Puchta, Nico J. R. van Eikema Hommes, Paul von R. Schleyer J. Am. Chem. Soc.; 2005; 127(8) pp 2425-2432 Abstract
↑ A Double-Twist Möbius-Aromatic Conformation of [14]Annulene Henry S. Rzepa Org. Lett.; 2005; 7(21) pp 4637
Abstract
↑ ab Renaissance of Annulene Chemistry E.R. Spitler, C.H. Johnson, M.M. Haley, Chemical Reviews; 2006; 106 pp 5344.
↑ ab The Annulenes F. Sondheimer, Accounts of Chemical Research; 1972; 5 pp 81.
↑ ‘’[10]Annulene: Bond Shifting and Conformational Mechanisms for Automerization’’ C. Castro, W. L. Karney, C. M. McShane, R. P. Pemberton, J. Org. Chemistry ‘’’ 2006’’’; 71 pp3001
↑ http://nptel.iitm.ac.in/courses/IIT-MADRAS/Engineering_Chemistry_III/pdf/1_Aromaticity.pdf
↑ Reduction and Oxidation of Annulenes K. Müllen, Chem. Rev. ‘’’1984’’’; 84. pp603
↑ F. Sondheimer, pure applied chemistry; 1963; 7 pp 363
↑ F. sondheimer, Proceedings of the Royal Society; 1967; 297 pp 173
↑ (1) Mills, W. H.; Nixon, I. G. J. Chem. Soc. 1930, 2510.
↑ Amnon Stanger J. Am. Chem. SOC. 1991, 113, 8277-8280
↑ Shogo Sakai, J. Phys. Chem. A, 2002, 106 (47), pp 11526–11532
↑ [1]
↑ ab [2]
↑ ab [3]
↑ ab [4]
↑ [5]
↑ [6]
↑ Carey, F. A. and Sundberg, R. J. Advanced Organic Chemistry: Part A — Structure and. Mechanisms, Fifth Edition, 2004.
↑ SOLOMONS & FRYHLE. Química orgânica, vol. 1, cap. 14 e 15. Oitava edição, 2005.
↑ Quim. Nova, Vol. 32, No. 7, 1871-1884, 2009
↑ CAREY, Francis A. Advanced organic chemistry: pt. A. Structure and mechanism. 5. ed. New York: Springer, 2007
↑ Koichi Komatsu and Toshikazu Kitagawa. Chem. Rev. 2003, 103, 1371−1427
↑ Lewis, G. N. J. Am. Chem. Soc. 1916, 38, 762-786
↑ Heitler, W.; London, F. Zeitschrift für Physik, 1927, 44, 455—472.
↑ Pauling, L.; Wheland, G. W. J. Chem. Phys. 1933, 1, 362.
↑ Carey, F. A.; Sundberg, R. J. Advanced Organic Chemistry Fourth Edition. Part A. Structure and Mecanisms. New York: Kluwer Academic / Plenum Publishers, 2000, 822p.
↑ Herndon, W. C.; Ellzey Jr., M. L. J. Am. Chem. Soc. 1974, 96, 6631
↑ Herndon, W. C. J. Mol. Struct. 1983, 103, 219.
↑ Randiê, M. J. Am. Chem. Soc. 1977, 99, 444
↑ Mulder, J. J. C.; Oosterhoff J. Chem. Commun. 1970, 305, 307.
↑ Kuwajima, S. J. Am. Chem. Soc. 1984, 106, 6496-6502.
↑ Tartadini, G. F.; Raimondi, M.; Simonetta, M. J. Am. Chem. Soc. 1977, 99-9, 2913-2918
↑ Li, S.; Jiang, Y. J. Am. Chem. Soc. 1995, 117, 8401-8406
↑ Breslow, R. Acc. Chem. Res., 1973, 6, 393
↑ Olah, G. A. J. Org. Chem. 2001, 66, 5943-5957
↑ ab Roberts, J. D.; Streitwieser Jr., A.; Regan, C. M. J. Am. Chem. Soc. 1952, 74, 4579-4582
↑ Breslow, R.; Chang, H. W.; Yager, W. A. J. Am. Chem. Soc. 1963, 85, 2033-2034
↑ Breslow, R.; Chang, H. W.; Hill, R.; Wasserman, E. J. Am. Chem. Soc, 1967, 89, 1112-1119
↑ Saunders, M. Berger, R.; Jaffe, A.; McBride, J. M.; O’Neill, J.; Breslow, R.; Hoffman, J. M.; Perchonock Jr. C.; Wasserman, E.; Hutton, R. S.; Kuck, V. J. J. Am. Chem. Soc. 1973, 95, 3017-3018
↑ Olah, G. A.; Prakash, G. K. S. Carbocations Chemistry. New Jersey: Wiley Interscience. 2004, Cap. 5 Antiaromaticity effects in cyclopentadienil carbocations and free radicals. pp. 103-123
↑ Melandra, J. L.; Mills, N. S.; Kadlececk, D. E.; Lowery, J. A.; J. Am. Chem. Soc. 1994, 116, 11622-11623
↑ Bushmarinov, I. S.; Lyssenko, K. A.; Antipin M. Y. Russian Chem. Rev. 2009, 78(4), 283-302. DOI: 10.1070/RC2009v078n04ABEH004017
↑ Camaroni, C. F.; Oliveira, K. T. Química Nova 2009, 32(7), 1871-1884.
↑ ab Firme, C. L.; Antunes, O.A.C.; Esteves, P.M. J. Braz. Chem. Soc. 2007, 18(7), 1397-1404.
↑ Cloonan, M. O. A new electronic theory of pericyclic chemistry and aromaticity is proposed: The Cplex-isoelectronic theory. Consistent with Santilli’s hadronic chemistry. International Journal of Hydrogen Energy 2007, 32, 159-171.
↑ SILVERSTEIN, R. M.; CLAYTON BASSLER, G.; MORRILL, T.C.; Identificação Espectrométrica de Compostos Orgânicos. 5ª edição. Rio de Janeiro: Guanabara Koogan, 1994.
↑ PAVIA, DONALD L.; LAMPMAN, GARY M.; KRIZ, GEORGE S.; Introdução à espectroscopia, 4ª edição, 2010.
↑ ALKORTA, I.; ELGUERO, J. New J. Chem., 1998, Pages 381-385.


