Cladograma

Cladograma horizontal, com o ancestral (sem nome) à esquerda.

Cladograma vertical, com o ancestral em cima.
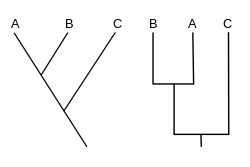
Dois cladogramas verticais, com o ancestral em baixo.

Exemplo de cladograma. No caso, destacando o grupo dos Dinosauria.
Cladograma é um diagrama usado em cladística que mostra as relações (filogenéticas ou genealógicas) entre táxons terminais, seja em nível de espécie ou grupos supra-específicos, formando grupos monofiléticos evidenciados por sinapomorfias, indicando uma história em comum, não necessariamente uma ancestralidade direta. Apesar de terem sido tradicionalmente obtidas principalmente por caracteres morfológicos, as sequências de DNA e RNA e a filogenética computacional são agora normalmente usados para gerar cladogramas.
Índice
1 Descrição
2 Etapas na construção de um cladograma
2.1 Determinação do grupo de estudo
3 Grupos externos
3.1 Seleção de caracteres
3.2 Homologias versus homoplasias
3.3 Plesiomorfias e apomorfias
3.4 Polarização
3.5 Matriz de caracteres
4 Geração de cladogramas
5 Seleção do melhor cladograma
6 Informações dos cladogramas
7 Referências
8 Bibliografia
9 Ligações externas
Descrição |
As ramificações de um cladograma representam as relações de ancestralidade, sendo um ramo, o ancestral do táxon (espécie, gênero, grupo artificial) que o segue. Os nós representam os eventos de cladogênese (divergência entre os táxons). A raiz representa o ancestral comum de todos os nós incluídos no cladograma. E por fim os nós terminais, que representam os táxons terminais, ou unidades taxonómicas funcionais (UTO), e sua distribuição na árvore constitui suas relações de semelhança e parentesco, o que caracteriza também a finalidade desse diagrama. Uma característica importante é que cladogramas mostram a relação entre grupos-irmãos, o tamanho dos ramos não correspondem a tempo de divergência (Dendogramas) ou quantidade de transformações morfológicas ou moleculares (Filogramas).
Cladogramas não são muito informativos se comparados a árvores filogenéticas, e não podem trazer detalhes sobre a evolução do grupo estudado, pois relacionados a processos de especiação, podem trazer padrões evolutivos distintos, por exemplo, os “nós” podem representar processos de brotamento (quando o ancestral dá origem a uma espécie descendente mas permanece vivente), divisão ou bifurcação (quando um ancestral origina duas espécies e se extingue, fusão ou hibridização (processo reticulado, com vários passos que não pode ser representado em árvores) ou ainda anagênese (na qual uma espécie dá lugar a sua descendente). Esses possíveis cenários evolutivos podem confundir a análise final do grupo estudado.
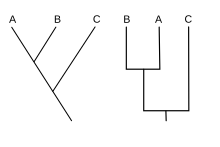
Dois exemplos de Cladogramas, mostrando que, independente da forma do diagrama, a posição ocupada pelos táxons terminais é o que realmente importa.
Etapas na construção de um cladograma |
Determinação do grupo de estudo |
O primeiro passo para produção de um cladograma é a escolha de um grupo de estudo. Este pode ser um táxon tradicional ou não. Por exemplo: uma família, onde os táxons terminais seriam gêneros. Ou poderia ser um gênero, sendo os táxons finais espécies. Os táxons terminais tampouco precisam restritamente ser dos mesmo nível taxonômico ou uma categoria tradicional (por exemplo os Bilateria, um táxon mais abrangente que inclui como táxons terminais Platyhelminthes, Nemertea, Mollusca, Sipuncula, Echiura e Metameria ("Annclida" + Ecdysozoa + Enterocoela) [1] . Mas é necessário que o grupo escolhido seja monofilético, e que essa monofilia seja sustentada por sinapomorfias. A monofilia do grupo já pode estar bem apoiada por uma boa lista de caracteres, entretanto, em casos especiais em que os estudos sobre o táxon ou grupo são recentes, é necessário um esforço maior, exigindo uma análise mais abrangente sobre suas relações para que possa ser inferida monofilia a esse grupo.
Um grupo monofilético é um conjunto de espécies incluindo uma ancestral e todas as suas espécies descendentes, ou seja, um conjunto de espécies que compartilha um ancestral comum mais recente. Antes do aperfeiçoamento do método filogenético, monofilas eram determinadas apenas por semelhanças, logo, espécies ou táxons supra-específicos eram unidos apenas por características morfológicas.
Um outro conceito é o de grupos merofiléticos, que correspondem a um grupo monofilético maior do qual foi retirado um grupo monofilético menor, ou seja, o que resta após a retirada de um ou mais táxons terminais. Apesar de não serem considerados bons agrupamentos, pois a distância entre os táxons é muito grande, alguns grupos merofiléticos foram mantidos, como Pisces.
Willi Hennig passou a classificar grupos merofiléticos em dois tipos: parafiléticos e polifiléticos, de acordo com as características que os unia. O primeiro seria diagnosticado por simplesiomorfias, e não possui todos os descendentes de um mesmo ancestral comum mais recente, como se um ou mais táxons terminais fossem retirados. E o segundo, seria unido por homoplasias, e incluiria no mínimo dois ancestrais comuns mais recentes. Esses agrupamentos não são muito aceitos, apesar de muitos ainda permanecerem na literatura por motivo didático.
Grupos externos |
São todos os organismos vivos exceto aqueles pertencentes ao seu grupo de estudo, e são determinados por polarização de caracteres. Um tipo importante é o grupo-irmão, que se caracteriza por ser o grupo externo filogeneticamente mais próximo do grupo de estudo, e o que fornece mais informações sobre o sentido da evolução dos caracteres. A relevância de se incluir grupos-externos a sua análise, é corroborar sua hipótese de monofilia ao grupo de interesse.
Seleção de caracteres |
Tendo os táxons terminais selecionados, chega o momento de descobrir e levantar dados de caracteres que variem dentro do grupo. Essa lista de características é o principal ponto na formação de um cladograma, pois é sobre elas que as hipóteses filogenéticas se apoiam. Geralmente matrizes de caracteres são criadas a partir de alguns pressupostos básicos, a saber: Cada espécie ou táxon supra-especifico tem dois estados de caráter, “0” é o plesiomórfico e “1” é apomórfico; Homoplasias, tanto por reversões ou convergências, são evitadas pelo princípio da parcimônia; E que um único passo, de plesiomórfico a uma condição mais derivada de estado, na grande maioria das vezes, representa uma mudança de uma espécie para outra, isso por que o processo evolutivo é discreto em cladogramas, para ser clara a distinção entre características e entre espécies.
Os caracteres utilizados podem ser de comportamento (nidificação, corte, cuidado parental, defesa de território, forrageio), embriologia, bioquímica, fisiologia, alimentação, morfologia (coloração, aspectos da cabeça, asa, morfometria óssea, etc).
Homologias versus homoplasias |

Homologia entre os ossos dos membros anteriores distais de mamíferos ("mãos").
No geral, estruturas homólogas são aquelas que indicam ancestralidade comum, ou seja, à luz da evolução, se duas espécies apresentam homologia em suas estruturas, quer dizer que seu ancestral comum também apresentava tal estrutura. Esta pode sofrer variações morfológicas e funcionais ao longo do tempo. Essa concepção passou a ser utilizada com o advento da biologia evolutiva, sendo que no período pré-Darwiniano, homologias caracterizavam um “plano da natureza”, num sentido sobrenatural [2] . Atualmente compõem a principal ferramenta utilizada pela Sistemática Filogenética.
De contrapartida, homoplasias são caracteres compartilhados entre duas ou mais espécies que não estavam presentes no ancestral comum entre elas, e que essa novidade teria surgido independentemente nas duas ou mais linhagens. Um exemplo muito comum são as asas de aves e de morcegos, que possuem a mesma função, mas tem origens diferentes. Em morcegos, a membrana alar estende-se entre os dedos de seus membros anteriores (homólogos ao braço humano), e em aves a membrana alar liga-se ao tórax pela parte distal da asa [3] ; Além disso asas de pássaros são cobertas por penas, enquanto de morcegos é coberta por pele ref> RIDLEY, M. 2003. Evolução. Porto Alegre. Artmed. 3ed. 752p. </ref> . Essas semelhanças são meramente superficiais, e surgiram mais de uma vez, provavelmente por que esses dois organismos sofreram pressões evolutivas muito parecidboas na aerodinâmica de suas asas (forma, tamanho, movimento) [4].

Relação homoplástica entre as asas de 1. Pterosauro (extinto), 2. Morcego e 3. Ave
Para cladística, a relevância dessa diferença se destaca no momento de inferência filogenética, no qual homoplasias não revelam relações de parentesco, enquanto homologias sim [5] .
Homoplasias causam muitas distorções na filogenia, pois podem ser confundidas, durante a análise, como sinapormofias, unindo duas linhagens, sendo que, na realidade, foram dois eventos independentes; o que caracteriza uma dificuldade imposta pela natureza evolutiva à sistemática. Durante a análise do cladograma, um índice é utilizado para mensurar a quantidade de eventos homoplásticos na árvore, ou para um caráter em particular. O Índice de Consistencia [6] é a razão entre o número mínimo de passos de uma série de transformação ou conjunto de séries de transformação pode exibir (m), pelo número efetivo de passos apresentado na evolução do caráter ou mostrados no cladograma (s). O valor de índice “1” equivale a nenhuma homoplasia, e quanto mais próximo do “0”, mais eventos homoplásticos são mensurados.
Plesiomorfias e apomorfias |
Em uma série de transformação, que seria a sequência de modificações que uma estrutura sofreu até sua condição mais derivada; a condição original (mais antiga) é denominada Plesiomorfia, enquanto a mais recente, derivada, é chamada de Apomorfia. Em sistemas naturais, essas condições são compartilhadas pelos indivíduos de uma ou mais espécies, nesse caso passam a ser chamados de Simplesiomorfia e Sinapomorfia. Assim, um dado estado de caráter é simplesiomorfico (ectotermia em répteis, anfíbios, outros vertebrados e invertebrados) para um grupo, ou sinapomórfico (presença de pêlos em mamíferos, celoma em celomados) para um grupo.
Séries de transformação lineares podem apresentar apenas duas condições, uma plesiomorfica e outra apomorfica (situação mais simples)(a presença de vértebras corresponde a uma condição apomorfica em relação a condição plesiomorfica “sem vertebras, por exemplo), ou ainda pode abranger condições intermediárias, na qual um estado sempre será apomórfico em relação ao estado a partir do qual se modificou (por exemplo, a modificação da condição tetrápode original de Amniota para a postura bípede dos humanos, na qual verifica-se várias etapas, com modificações sucessivas na inclinação da coluna e na forma de outros ossos). Vale ressaltar, que nesses casos, uma modificação anterior estará presente na posterior nas mesmas estruturas, ou seja, no caso do exemplo da condição bípede em humanos, uma novidade foi da condição tetrápode para um semi-ereta (a -> b), e depois da condição semi-ereta para a totalmente ereta (b -> c). A apomorfia quadrúpede -> semi-ereto vai estar tanto em organismos semi-eretos quanto naqueles totalmente eretos [7]. Existem ainda series com eventos de bifurcação, nas quais de uma mesma condição plesiomórfica surge duas ou mais condições apomórficas.
Só existem sinplesiomorfias quando houve mudanças nos estados de caracteres para uma condição apomórfica, caso contrário, essa condição antiga pertencente ao grupo denomina-se Arqueomorfia (por exemplo a coluna vertebral em Vertebrata, ainda não existe nenhuma condição derivada a esta). Outro conceito é o de Autapomorfia, trata-se de uma apomorfia para um único ramo terminal de um cladograma; Ou seja, caso especial onde uma sinapomorfia é compartilhada apenas pelo táxon terminal. Contudo, se for feito um estudo sobre esse táxon em particular, de uma forma menos abrangente, essa autapomorfia será apenas uma sinapomorfia para o grupo.
Polarização |
É o método pelo qual reconhecemos o que é plesiomórfico e o que é apomórfico, ou seja a polaridade do caráter, e como esses estados de caracteres serão dispostos no cladograma. Existem basicamente dois meios de determinar o sentido da mudança: método do grupo-externo e por registro fóssil.
O primeiro utiliza um grupo-externo relacionado ao grupo de estudo para determinar qual caráter é ancestral. Geralmente o grupo-externo apresenta tal característica. Por exemplo, Amniota é o grupo formado por répteis, aves e mamíferos; todos esses animais possuem uma membrana do ovo, chamado âmnio, durante o seu desenvolvimento. Sabe-se que amniotas são um grupo monofilético, ou seja, eles todos compartilham um mesmo ancestral comum. Pegando um conjunto de seis espécies amnióticas como um rato, um canguru, um pássaro do paraíso, um pisco, um crocodilo e uma tartaruga, onde duas são vivíparas (rato e canguru) e o restante é ovípara, como saber qual caráter é apomórfico? Com isso é selecionado um grupo-externo não amniota próximo filogeneticamente, como salamandras, sapos ou um peixe. Nesses grupos oviparidade predomina, consequentemente essa característica é considerada basal (plesiomórfica) em relação á viviparidade (apomórfica). Ou seja, cangurus e ratos seriam táxons mais derivados dentro do grupo de estudo [8] . Entretanto esse método tem ressalvas, pois um determinado grupo externo pode sugerir que um caráter é basal, mas ou outro grupo externo pode apontar um outro caráter como basal. Assim é necessária uma análise com o máximo de grupos ou espécies próximas relacionadas que corroborem a mesma polaridade.
Outro ponto é o fato de ser necessário um conhecimento prévio da monofilia do grupo-interno e de seus possíveis grupos-irmãos comparáveis [9] . Caso o estudo seja feito com grupos na qual sua monofilia não esteja bem embasada, acarretará em uma complicação na análise, com confusão nos dados que só seria solucionado tomando o estudo do ponto inicial. Com isso, é recomendado que antes de se iniciar o estudo, sejam feitas as relações de parentesco em um nível mais abrangente, mesmo que de uma forma superficial, delimitando um grupo que seja monofilético, para depois iniciar a análise com os elementos que fazem parte desse grupo [10].
O método de polarização utilizando fósseis é ainda mais simples. Nesse caso caracteres deixados antes no registro fósseis serão plesiomórficos em detrimento de fósseis mais recentes, que apresentariam caráter derivado. Isso funciona muito bem para a evolução de mamíferos a partir de répteis-tipo-mamíferos, devido ao vasto registro fóssil encontrado. Entretanto, se uma condição apomórfica se preservar antes do que sua condição basal, em registros fosseis restritos, a interpretação paleontológica será o contrário da realidade.
O desenvolvimento embriológico também pode ser utilizado como método de polarização de caracteres.
Matriz de caracteres |
As matrizes são os depósitos das informações reunidas até agora. Exibem em cada ponto a condição de um caráter em um determinado táxon. Elas correspondem à base de dados sobre a qual os cladogramas serão construídos. Erros na matriz de caracteres corresponde a erros de topologia na árvore, ou seja, cladogramas falsos [11] .
Tecnicamente, a maneira de orientar as matrizes em colunas e em linhas pode variar, mas normalmente são feitas colocando os táxons nas linhas, e os caracteres nas colunas. Em relação à ordenação de caracteres e táxons na matriz, se já houver alguma inferência sobre a filogenia do grupo, esta pode ser utilizada para organizar os dados na matriz, no qual a sequência dos táxons podem seguir a hierarquia da classificação tradicional. À medida que o estudo prossegue, novas sinapomorfias encontradas podem unir grupos monofiléticos, sendo necessária uma nova matriz. Um método de organizar os táxons por ordem alfabética, por exemplo, pode dificultar a visualização de caracteres compartilhados e de homoplasias.
Existem três tipos de matrizes:
Matriz pictórica: apresenta desenhos dos caracteres ordenados em colunas para melhor comparação entre os táxons; sendo muito importante no momento de levantamento de dados. São relevantes na detecção de caracteres em estruturas complexas, com diversas mudanças independentes em várias partes (como genitália e apêndices de insetos).
Matriz não-polarizada: estão relacionadas, assim como as matrizes pictóricas, ao início da coleta de caracteres. Incluem valores numérico, variação de tamanho, um pequeno desenho, descrição de cor, sinal indicando presença (+) ou ausência (-), e assim por diante. São úteis para visualizar caracteres com mais de um passo de transformação. E são puramente descritivas, usada para levantamento de dados, não indicando plesiomorfias ou apomorfias.
Matriz polarizada: é a matriz que realmente fornece dados para a construção de cladogramas. Sua lista de caracteres (descrição das condições de estruturas e suas variações dentro do grupo estudado) informa o que foi interpretado pelo autor como plesiomórfico e apomórfico para cada caráter. Sinapomorfias e homoplasias só serão identificadas no momento em que esse conjunto de dados da matriz passa para um cladograma, obedecendo os critérios da parcimônia. É sobre essa lista que as hipóteses filogenéticas se baseiam, sendo de suma importância. Matrizes polarizadas contêm as informações das matrizes não-polarizadas sob a luz das séries de transformação e polarização, e podem ser mais de uma em caso de estados não-lineares (séries de transformação bifurcadas).
No decorrer da análise, podem surgir casos especiais de caráteres não comparáveis, ou seja, a inexistência de informações sobre alguns componentes do grupo de estudo, como táxons terminais ou grupos-externos. Pode ser um evento de má qualidade do material para análise, como partes perdidas ou quebradas, fósseis incompletos; ou ainda se o caractere for fisiológico ou comportamental, e você possui apenas peças fixadas em museus; ou o caráter pertence a estruturas inexistentes em outras espécies, ou a estruturas que ainda não são documentadas na literatura; E ainda um caso específico para espécies partenogenéticas, nas quais inexistem machos.
Geração de cladogramas |
Quando considerados poucos táxons, essa etapa pode ser feita manualmente. Entretanto, se a análise inclui muitos táxons e dezenas de caracteres, é necessário o auxílio de um programa computacional. No primeiro caso, caracteres da matriz polarizada são adicionados um a um em uma politomia. À medida que a adição prossegue, hipóteses de monofilias são geradas, tomando cada caráter como apomórfico. A ordem de adição de caracteres não é relevante quando sua matriz polarizada não apresenta incongruências, ou seja, carateres diferentes indicam que o grupo ou espécie pertencem a grupos monofiléticos diferentes, podendo representar homoplasias ou reversões, o que não é bom para análises cladísticas que prezam o princípio da parcimônia. Por outro lado, congruências são apomorfias que unem grupos monofiléticos. É válido ressaltar que a construção deficiente dos caracteres (determinação de homologias, definição de estados de caráter, polarização, ordenação e séries de transformação) leva a incongruências, logo à construção de cladogramas ruins, falsos.
O número de cladogramas formados em uma análise é proporcional ao número de táxons incluídos, e cresce exponencialmente [12] . Assim, para um matriz com 3 táxons, existem 3 possibilidades de árvore, com 5 táxons, 15 possibilidades; com 10 táxons chega a 34459425 possibilidades. Nesse caso, programas de computador são úteis como ferramentas para encontrar a árvore mais parcimoniosa, utilizando algorítimos específicos.
A busca pela árvore mais parcimoniosa é uma tentativa de otimizar a matriz de caracteres em uma dada topologia, ou seja, a árvore que melhor expressa a matriz de dados. Aqui, dois conceitos devem ser introduzidos, o de parcimônia e de otimização.
Otimização é o procedimento para se construir o cladograma mais parcimonioso, com menos passos evolutivos [13] . Por isso os procedimentos de escolha de caracteres até sua manipulação (polarização, series de transformação, ordenação e plotagem na matriz) devem ser feitas com atenção e olhar crítico.

Dois eventos diferentes de distribuição de caracteres. O Cladograma mais superior da imagem, representa os táxons 1 e 2 como um grupo monofilético, pelo aparecimento unico co caráter "B". E abaixo, um Cladograma com um cenário diferente, no qual o caráter "B" teria surgido duas vezes, fazendo com que 1 e 2 fossem parafiléticos. O primeiro caso é o cenário bom, já que apresenta maior Parcimônia.
A parcimônia refere-se ao princípio que a filogenia com menor número de passos evolutivos possíveis é a melhor estimativa das verdadeiras relações filogenéticas. Isso é largamente aceito pois a evolução de um caráter é improvável, então ele tende a permanecer o mesmo, portanto, uma mudança seguida de uma reversão ao caráter antigo é improvável, visto que mudanças necessitam de mutações genéticas, neutras ou não, o que são eventos igualmente improváveis. Logo as características tendem a ser as mesmas ao longo das gerações. Isso é corroborado pela relação prole-progenitor, onde há transmissão de caracteres. Existem programas de computador próprios para buscar a árvore mais parcimoniosa, exemplos são Hennih86 e PAUP, os mais utilizados.
Além da parcimônia, outros fatores são utilizados no momento de escolher a melhor árvore. Quando se obtém árvores parcimoniosas, com o mesmo número de passos evolutivos e topografia, dois critérios algorítmicos podem ser tomados para escolha do cladograma com a melhor hipótese: Acctran e Deltran (swofford & maddison, 1987). Acctran (procedures that ACCelerate the evolutionary TRAnsformation of a character) atribui a origem de um caráter a um nível mais abrangente, privilegiando uma origem anterior de um caráter seguido por uma reversão. Já Deltran (procedures that DELays the evolutionary TRANsformation of a character) privilegia o aparecimento mais tardio de condições idênticas (homoplasias). A explicação da decisão entre homoplasias ou reversões deve ser muito clara pelo autor, usando os cárteres como principais argumentos [14] . Levando em consideração as mudanças em nível de DNA, uma aquisição de uma estrutura, com uma aquisição e uma perda (reversão) é mais provável do que duas aquisições (homoplasia). E em casos onde a apomorfia corresponde a uma perda, a probabilidade de dois eventos de perda independentes (homoplasia) é maior do que uma perda e uma reaquisição secundária (reversão).
Seleção do melhor cladograma |
A análise de parcimônia pode gerar inúmeras árvores mais parcimoniosas, o que pode ser chamado de “floresta” [15] , e é difícil escolher qual delas melhor infere as relações filogenéticas do grupo. Para isso, existem métodos de consenso de árvores, que buscam otimizar a topologia dos cladogramas (não a matriz de caracteres):
Consenso estrito: o mais clássico da cladística, apresentado por Sokal & Rohlf (1981) [16] . Método mais conservativo, que garante a congruência de todas as árvores parcimoniosas propostas; Entretanto apresenta politomias, o que não é considerado um bom resultado pelos sistematas. Mas pode trazer informações construtivas no sentido da sua qualidade de matriz de dados.
Consenso semi-estrito ou dos componentes combináveis [17] : Derivação do consenso estrito, que preserva todas as informações possíveis de cada árvore original, desde que não apresentem contradições. No caso de dois cladogramas, onde um apresenta uma politomia e o outro dicotomias resolvidas, o consenso semi-estrito preservará as relações resolvidas na árvore de consenso. Caso não haja politomias, o resultado será semelhante ao do consenso estrito.
Consenso de Adams [18] : Preserva o máximo possível da resolução das árvores da floresta original, mesmo que a árvore consenso seja totalmente diferente de qualquer uma das originais, mascarando os resultados (ocultando grupos monofiléticos formados e aceitando grupos merofiléticos). Não é aceito em analises de parcimônia.
Consenso de maioria [19] : aceita a resolução que mais aparece entre as árvores originais, ou seja a maioria, com o valor limite de 50% dos cladogramas originais. A topologia produzida não é congruente com a floresta original, e apresenta valores numéricos associados aos clados (como a porcentagem de árvores originais está representada ali).
Outros algorítimos para otimização de cladogramas são os de Máxima verossimilhança, de Inferência Bayesiana, Método dos mínimos quadrados, parcimônia e outros. No geral, esses algoritmos compara as árvores geradas umas em relação às outras, e por fim encontra uma que é a melhor alternativa. [20]
Informações dos cladogramas |
Basicamente, cladogramas contêm hipóteses sobre a filogenia de um determinado grupo. Essa hipótese pode ser verdadeira ou falsa dependendo da interpretação dos dados considerados para o estudo. Correspondem ainda a um conjunto de informações sobre monofilia, ou seja, caso a monofilia de um subgrupo seja refutada, isso não necessariamente invalida as demais. Erros de analise podem envolver casos de afirmação incorreta de homologia primária de caracteres, ou sobre a polaridade desses caracteres; Afirmações erradas sobre homologias secundárias (apomorfias compartilhadas sendo homoplasias ao invés de sinapomorfias, ou ainda se as sinplesiomorfias não são realmente eventos de reversão); E equívoco no momento de inferir monofilia ao táxon de estudo.
As informações apresentadas em um Cladogramas podem sofrer transformações à medida que o conhecimento sobre os táxons e sua diversidade aumentam, com a adição de dados de espécies ou caracteres; ou mudança na polarização, e na monofilia do grupo, por exemplo, podendo ou não mudar a topologia da árvore.
Por fim, um bom cladograma é uma ferramenta muito útil nas áreas biológicas como um todo, devido sua capacidade de retrovisão dos eventos que ocorreram na evolução de um grupo.
Referências
↑ AMORIN, D. S. Fundamentos de Sistemática Filogenética. Holos. São Paulo. Edição única. 154p.
↑ RIDLEY, M. 2003. Evolução. Porto Alegre. Artmed. 3ed. 752p.
↑ AMORIN, D. S. Fundamentos de Sistemática Filogenética. Holos. São Paulo. Edição única. 154p.
↑ GABORA, L. Convergent Evolution. Acesso Abril de 2013
↑ RIDLEY, M. 2003. Evolução. Porto Alegre. Artmed. 3ed. 752p.
↑ KLUGE, A.G. & FARRIS, J.S. 1969. Quantitative phyletics and the evolution of anurans. Syst. Zool. 18: 1-32
↑ AMORIN, D. S. Fundamentos de Sistemática Filogenética. Holos. São Paulo. Edição única. 154p.
↑ RIDLEY, M. 2003. Evolução. Porto Alegre. Artmed. 3ed. 752p.
↑ RIDLEY, M. 2003. Evolução. Porto Alegre. Artmed. 3ed. 752p.
↑ AMORIN, D. S. Fundamentos de Sistemática Filogenética. Holos. São Paulo. Edição única. 154p.
↑ AMORIN, D. S. Fundamentos de Sistemática Filogenética. Holos. São Paulo. Edição única. 154p.
↑ FERRAREZZI, H. & MARQUES, A. C. Procedimentos de Análise em Cladística Numérica. Acesso em Abril de 2013
↑ KITCHING, J.I. et al. Cladistics: The Theory and Practice of Parsimony Analysis. 1998. Oxford University Press. 2ed. 228p.
↑ AMORIN, D. S. Fundamentos de Sistemática Filogenética. Holos. São Paulo. Edição única. 154p.
↑ FERRAREZZI, H. & MARQUES, A. C. Procedimentos de Análise em Cladística Numérica. Asesso em Abril de 2013
↑ SOKAL, R.R. & ROHLF, F.J. 1981. Taxonimic congruence in the Lepopodomorpha reexamined. Syst. Zool., 30: 309-325.
↑ BREMER, K. 1990. Combinable componente consensus. Cladistics, 6: 369-372.
↑ ADAMS, E.N. 1972. Consensus techniques and the comparison of taxonomic trees. Syst. Zool.,37: 27-29.
↑ MARGUSH, T. & McMORRIS, F.R. 1981. Consensus n-trees. Bull. Math. Biol. 43: 239-244
↑ RIDLEY, M. 2003. Evolução. Porto Alegre. Artmed. 3ed. 752p.
Bibliografia |
- RIDLEY, M. 2003. Evolução. Porto Alegre. Artmed. 3ed. 752p.
- FERRAREZZI, H. & MARQUES, A. C. Procedimentos de Análise em Cladística Numérica. Asesso em Abril de 2013.
- GABORA, L. Convergent Evolution. Acesso Abril de 2013.
- AMORIN, D. S. Fundamentos de Sistemática Filogenética. Holos. São Paulo. Edição única. 154p.
- MARGUSH, T. & McMORRIS, F.R. 1981. Consensus n-trees. Bull. Math. Biol. 43: 239-244.
- ADAMS, E.N. 1972. Consensus techniques and the comparison of taxonomic trees. Syst. Zool.,37: 27-29.
- BREMER, K. 1990. Combinable componente consensus. Cladistics, 6: 369-372.
- SOKAL, R.R. & ROHLF, F.J. 1981. Taxonimic congruence in the Lepopodomorpha reexamined. Syst. Zool., 30: 309-325.
Ligações externas |
 Conteúdo relacionado com Cladograms no Wikimedia Commons
Conteúdo relacionado com Cladograms no Wikimedia Commons


